INTRODUCTION
Despite recent advances in ablative surgical resection, radiotherapy, and chemotherapy, recurrence and metastasis remains significant problems in the treatment of head and neck squamous cell carcinoma (HNSCC). Local or regional disease recurs in 30% of advanced HNSCC patients, and distant metastases appear in 25% of patients, leading to a 5-year survival rate of only 40% [1]. For these reasons, development of novel therapeutic strategies for head and neck cancers could lead to significant improvements in survival.
5-Fluorouracil (5-FU) is a major anti-tumor chemotherapeutic agent that has been widely used in the treatment of head and neck cancers alone or in combination with other chemotherapeutic agents [2]. However, this widely-used anticancer agent has several limitations, including serious side effects, such as pancytopenia and diarrhea, anorexia, nausea, and taste changes, and high dose levels are required to achieve a response. High systemic toxicity of 5-FU could possibly be circumvented by introducing directed expression of bacterial and/or yeast cytosine deaminase (CD), an enzyme that converts the far less toxic substrate 5-fluorocytosine (5-FC) into 5-FU [3]. 5-FU is capable of non-facilitated diffusion in and out of cells, resulting in a significant bystander effect caused by CD/5-FC treatment [4].
Successful gene therapy technology relies on the delivery of the therapeutic product to the appropriate target cells. Introduction of the transgene of interest into stem cell types poses an attractive cell-based delivery strategy [5]. The discovery of the inherent tumor-tropic properties of neural stem cells (NSCs) could serve as a novel adjuvant strategy to complement current head and neck cancer treatments. Recent studies have shown that NSCs can be used to target therapeutic genes to brain tumors, including malignant glioma [6] and melanoma brain metastases [7]. We have expanded on these investigations to determine whether NSCs are also capable of targeting head and neck carcinomas in an animal xenograft model. Therapeutic proof-of-concept studies were performed using CD-expressing NSCs and systemic 5-FC prodrug administration.
MATERIALS AND METHODS
Cell culture
HB1.F3 (F3) is an immortalized human NSC line. It was derived from human fetal brain (ventricular zone) at 15 weeks of gestation and immortalized using an amphotropic, replication-incompetent retroviral vector containing v-myc [8-11]. This is a well-established and well-characterized NSC line that is multipotent, migratory, and non-tumorigenic in vivo [8-15]. F3 cells were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 2 mmol/L L glutamine, 100 units/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin B (Invitrogen, Grand Island, NY, USA).
The human head and neck cancer cell line SNU-1041 was maintained in DMEM containing 10% FBS, 100 units/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin B (Invitrogen).
Engineering of the human NSC line HB1.F3-CD
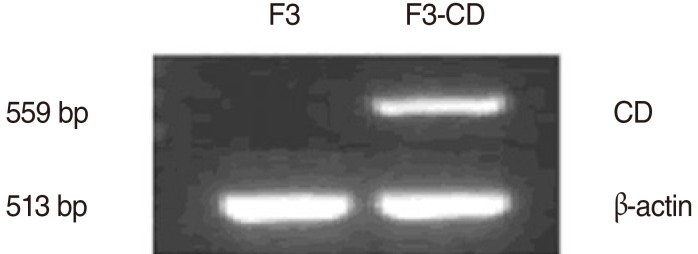
The clonal HB1.F3-CD (F3-CD) cell line was derived from the parental F3 line. An expression plasmid encoding Escherichia coli CD was constructed using the retroviral pBabePuro backbone and the 1.5 kb CD cDNA. Vectors were packaged by co transfection of pA317 cells with the CD-Puro plasmid and the MV12 envelope-coding plasmid. CD-Puro retroviral supernatant was used for multiple infections of F3 cells. Transduced F3-CD cells were selected in the presence of 3 µg/mL puromycin (Invitrogen) over 4 weeks. Successful transduction of the F3-CD cells was confirmed by reverse transcription PCR (rt-PCR) using the following primer pair: sense, 5'GCGCGAGTCACCGCCAGCCACACCACGGCGCGCGAGTCACCGCCAGCCACACCACGGC-3' and antisense, 5'GTTTGTAATCGATGGCTTCTGGCTGC-3'.
In vitro therapeutic efficacy of F3-CD
F3 cells, F3-CD cells, SNU-1041 cells (3×104 cells per well), and F3-CD cells (104 per well) co-cultured with SNU-1041 cells (2×104 per well) were plated in 96-well plates in triplicate and incubated overnight at 37℃. Culture medium was replaced with medium containing 0-2.5 mg/mL 5-FC after 24 hours. Four days later, plates were subjected to a standard MTT assay. Results were expressed as the percentage of proliferation and were normalized to proliferation of cells in culture medium with no 5-FC.
Labeling of F3-CD cells with ferumoxides
Ferumoxides, a superparamagnetic iron oxide contrast agent (Feridex, Berlex, Wayne, NJ, USA), was used for magnetic labeling of F3-CD. Feridex is FDA-approved for clinical use in liver imaging and is commercially available [16]. Ferumoxides acts by reducing the transverse relaxation time (T2) on T2-weighted magnetic resonance imaging scans, causing labeled cells to appear as areas of reduced signal intensity.
For labeling experiments, ferumoxides (25 mg/mL) and poly-L-lysine (PLL, Sigma, St. Louis, MO, USA; 0.75 mg/mL) were mixed together with media and incubated at room temperature for 60 minutes [17,18]. PLL acts as a transfection reagent by binding the dextran-coated ferumoxides nano-particles via electrostatic interactions [19] and facilitating uptake into cells through membrane destabilization [20]. The F3-CD cells were then incubated for 24 hours at 37℃ to allow uptake of ferumoxides into the cells. Labeled F3-CD cells were identified as blue dots using Prussian blue staining.
In vivo visualization of migration of F3-CD cells to the tumor site
Six-week-old female athymic nude mice (BALB/c-nu/nu) were used for in vivo experiments in accordance with institutional guidelines under approved protocols. A suspension of 1×106 SNU-1041 cancer cells in 100 µL phosphate-buffered saline (PBS) was administered by subcutaneous injection (s.c.) at the posterior neck of nine animals. On day 7, 1×106 F3-CD cells tagged with Feridex were injected into the tumor-bearing mice using one of three different methods: at the tumor center (n=3), 1.5 cm from the tumor margin (peritumoral injection, n=3), or intravenously (i.v.) in the tail vein (n=3). To detect F3-CD cells located near tumor tissues, animals were sacrificed on day 14, and Prussian blue staining was performed on tissues from the brain, lung, liver, spleen, heart, and tumor.
In vivo therapeutic efficacy of F3-CD cell-mediated treatment
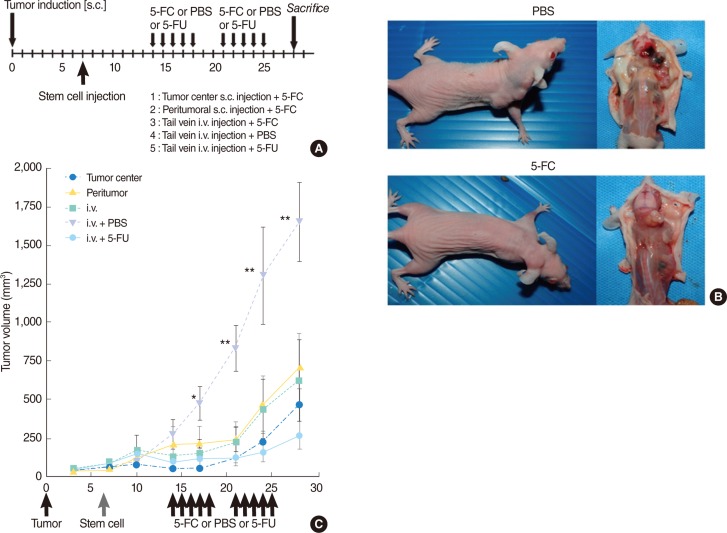
Six-week-old female athymic nude mice (BALB/c-nu/nu) were used. Cancer cells were administered as a suspension of 1×106 SNU-1041 cells in 100 µL PBS s.c. at the posterior neck in 50 animals. On day 7, 1×106 F3-CD cells in 100 µL PBS were injected by one of three different methods: at the tumor center (group 1, n=10), 1.5 cm from the tumor margin (group 2, n=10), or i.v. in the tail vein (groups 3-5, n=30).
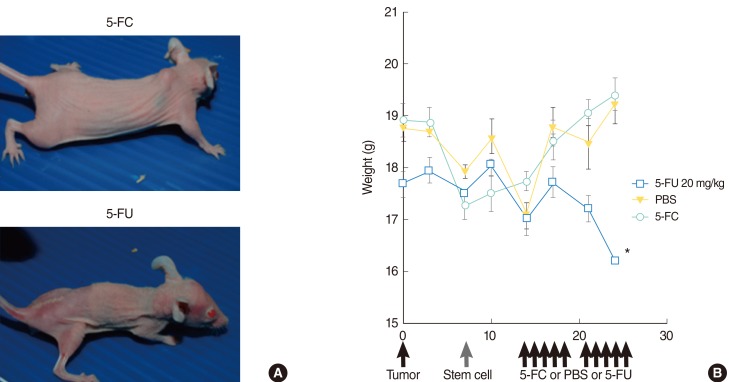
Beginning at day 14, all animals in groups 1, 2, and 3 were treated with 500 mg/kg/d 5-FC diluted in PBS, delivered by intraperitoneal injection (i.p.) in two rounds of five consecutive days with a 2-day break where indicated. Group 4 (control group) animals were treated only with PBS, and group 5 animals were treated with 20 mg/kg/d 5-FU i.p., instead of 5-FC.
Tumors were measured using calipers, and tumor volume was calculated according to the following formula: volume=(height×width2)/2 [21]. Animals were weighed daily. At the end point of the experiment, animals were sacrificed and tumors were excised. Results were evaluated as mean tumor volume or weight±standard error (SE).
RESULTS
Confirmation of CD production in F3-CD cells
Expression of the CD transcript in F3-CD cells was confirmed by rt-PCR. Results showed that the CD transcript was expressed only in F3-CD cells, not in the parental F3 cell line (Fig. 1).
In vitro therapeutic efficacy of F3-CD cell-mediated therapy
To confirm the "bystander effect" caused by release of 5-FU from F3-CD cells, cell viability studies were performed. Growth of 5-FC-treated, F3-CD cells, and SNU-1041 cells co-cultured with F3-CD cells was significantly inhibited in a dose-dependent manner, but growth of F3 cells or SNU-1041 cells alone was unaffected by 5-FC treatment (Fig. 2). SNU-1041 cells cultured with F3-CD cells exhibited no growth inhibition in the absence of 5-FC treatment. These observations indicate that F3-CD cells can convert sufficient amounts of 5-FC to 5-FU, which diffuses locally to effectively kill SNU-1041 cells in vitro.
In vivo visualization of migration of F3-CD cells to tumors
F3-CD cells were identified as blue dots stained with Prussian blue and were found in tissues taken from tumors, regardless of the method of injection. Most of the stained cells were found surrounding the tumor border. The density of stem cells was highest in the tumor center injection group and lowest in the tail vein injection group (Fig. 3A-C).
Tissues from the brain (Fig. 3D), heart (Fig. 3E), kidney (Fig. 3F), lung, liver (Fig. 3H), and spleen (Fig. 3I) were negative for Prussian blue staining in the tumor center injection group and the peritumoral injection group. Tissue from the lung (Fig. 3G) exhibited minimal staining in the tail vein injection group. Other tissues from the tail vein injection group were negative for Prussian blue staining, indicating an absence of F3-CD cells in these tissues.
In vivo therapeutic efficacy of F3-CD cell-mediated treatment
In vivo studies showed a significant reduction in tumor volume in 5-FC-treated animals (groups 1-3) in comparison with control animals treated with PBS (group 4; P<0.01) (Fig. 4). Use of different injection methods did not lead to significant changes in tumor volume. No significant difference in tumor volume was observed between 5-FC-treated animals injected with F3-CD cells (groups 1-3) and animals treated with 5-FU (group 5).
Side effects of treatment
There was no significant difference in weight between animals treated with 5-FC and animals treated with PBS. Animals treated with 5-FU showed significant weight loss in comparison to animals treated with 5-FC (Fig. 5). Animals treated with 5-FU also exhibited bloody stools.
DISCUSSION
In this study, we tested the hypothesis that NSCs expressing a therapeutic gene would have positive therapeutic effects on HNSCC. We chose the human F3 NSC cell line, because this human stem cell line is well-established and well-characterized [8-15]. The F3 human NSC line was immortalized using v-myc [8,10,11] and is an ideal vehicle for the delivery of suicide genes or immunomodulatory genes to tumor targets, including primary brain tumors and brain metastases [22,23]. The present study found no abnormalities caused by treatment with F3.CD cells; however, tumor formation must be considered a risk, due to the presence of the v-myc oncogene in the F3 human NSCs. In this study, no sign of local or systemic toxicity was observed in the F3-CD treated groups, although the therapeutic effects were assessed 3 weeks after NSC injection.
The in vitro and in vivo studies in this report show for the first time the ability of NSCs to target HNSCC, indicating that tumor tropism of NSCs is not limited to brain tumors. A possible mechanism of preferential stem cell homing and engraftment into the tumor mass could be attributed to the necessity for tumor stroma formation during the process of tumor growth, which includes tissue remodeling and involves high levels of stem cell proliferation [24]. Migratory properties must be further evaluated to determine tumor type specificity and to identify signaling molecules involved in this process, because of its relevance to potential therapeutic applications. It has previously been reported that epidermal growth factor, platelet-derived growth factor, SCF/c-Kit, stromal cell derived factor-1/CXCR4, vascular endothelial growth factor (VEGF), VEGF receptor 1 (VEGFR1), and VEGFR2 may play roles in these intrinsic tumor-specific migration capabilities and interactions [25,26].
After confirmation of the tropism of NSCs for head and neck squamous carcinoma cells, we tested their therapeutic potential using the CD enzyme/5-FC prodrug system. CD is a bacterial enzyme that converts the non-toxic prodrug 5-FC into the cytotoxic drug 5-FU, a nucleotide analog that disrupts DNA synthesis in proliferating cells [27]. Because mammalian cells do not express significant amounts of CD, as shown in the present study, 5-FC is not toxic at concentrations that result in strong antimicrobial activity. Thus, tumor cells transfected with the E. coli CD gene become selectively sensitive to the toxic effects of 5-FC. For example, expression of E. coli CD in glioma cells confers lethal sensitivity to systemic administration of 5-FC [28]. Furthermore, the enzyme/prodrug system also has a bystander effect [29], shown by the death of unmodified tumor cells adjacent to genetically modified cells. NSCs engineered to express a suicide gene like CD are a particularly beneficial therapeutic choice because they can generate agents that both kill tumor cells and result in self-elimination upon initiation of cell division in vivo.
Potential cancer therapies involving selective activation of prodrugs in tumor tissues by exogenous enzymes have used several distinct mechanisms, including gene directed enzyme prodrug therapy, virus-directed enzyme prodrug therapy, and antibody-directed enzyme prodrug therapy [30]. The cell-directed enzyme prodrug therapy using NSCs described in this report is potentially more flexible than previous methods, because of the tumor-tropic capacity of NSCs. Because of an impressive bystander effect, as little as 2% of the tumor mass containing CD-expressing cells may generate significant oncolysis [29]. This unique property increases the likelihood of achieving therapeutic efficacy. For clinical application of cell-directed enzyme prodrug therapy, the interval between enzyme and prodrug administration should be optimized so that prodrug delivery occurs only after NSCs localize to tumor site to avoid systemic toxicity.
Although a significant reduction in tumor volume was induced by cell-based therapy in this study, none of the animals remained tumor-free or was cured in this experimental setting. It is generally acknowledged that a successful long-term effective therapy or curative approach would require a combination of multiple treatment strategies. In our approach, the 5-FU produced at the site of tumor formation would be expected to eliminate the rapidly proliferating tumor cell population, based on the mechanism of action of 5-FU [4]. However, quiescent and non-dividing tumor cells present within the tumor mass may be able to withstand the toxicity. Our future studies will be directed towards optimization of protocols for dosage and timing of the therapy to achieve maximum suppression of tumor growth. As the therapeutic benefit from one time drug delivery may be short-lived as the tumor regenerates, additional cycles of F3-CD and 5-FC therapy could be given to eradicate proliferating cancer cells not removed by the first wave of therapy. Alternatively, NSCs could be engineered to express other potentially beneficial therapeutic genes (e.g., differentiating agents, immune enhancing agents, apoptosis-promoting agents, antiangiogenic agents), which could be used sequentially or in combination with F3-CD treatment.
In the clinical setting, NSC therapy could be used in conjunction with standard therapies in the near future. Particularly in the case of recurrence with tumor dissemination, NSCs could be injected into veins to target residual invading and disseminating tumor cells. Furthermore, based on our study, the same approach could be exploited using other sources of stem cells and they could be engineered to deliver other antitumor agents for cancer therapies.
In conclusion, this study indicates that NSCs have migratory capacity and exhibit tropism for HNSCC. Furthermore, these cells can be genetically modified ex vivo to express genes that have therapeutic efficacy against HNSCC. These results suggest the potential utility of NSCs as a targeted therapeutic delivery vehicle for the treatment of HNSCC.