INTRODUCTION
Allergic rhinitis (AR), an immunoglobulin E (Ig E)ŌĆöand T helper (Th)2-mediated inflammatory nasal disease, is caused by sensitized immune responses to inhaled allergens such as pollens, environmental fungi, dust mites, bacteria, and animal dander. This allergen-specific immune response is thought to arise from an imbalance in Th1ŌĆōTh2 immune regulation that results in increased levels of Th2 cytokines [1,2]. Nasal epithelial cells exposed to external allergens induce Th2 inflammatory responses that then spread to the upper airway mucosa [3,4]. The allergen-mediated inflammatory immune response begins with increased secretion of epithelial cellŌĆōderived cytokines, and the process of allergen sensitization involves the participation of antigen-presenting cells, T lymphocytes, and B lymphocytes and depends on environmental allergen exposure. Sensitization results in generation of allergen-specific IgE, which circulates in the peripheral blood and attaches itself to the surface of all mast cells and basophils, including those of the nasal mucosa [1,2]. Subsequent nasal exposure to allergen activates these cells, and the release of classic mediators of the allergic reaction triggers acute nasal symptoms. Therefore, the nasal epithelium might be responsible for the vast majority of allergic inflammation in response to inhaled allergens, and research on the regulation of epithelial cellŌĆōderived cytokines is needed to develop a more effective approach to treat AR [4,5].
Human mucosal surfaces are in direct contact with the external environment and are susceptible to invasion and colonization by various allergens and pathogens [6]. The respiratory mucosa is constantly exposed to inhaled pathogens and allergens that directly impact mucosal immune mechanisms [7]. Studies focusing on interactions between the mucosal microbiome and the host have increasingly considered the contribution of mucosal immune responses and specific microbiome-mediated protection from external pathogens to integrating environmental signals [7]. The most commonly studied symbiont microbiota are those of the intestinal mucosa, which are essential for intestinal immune responses in conditions of both health and disease, and dysbiosis of the intestinal mucosal microbiota and disruption of its close interactions with the host can destroy this steady-state immune balance in concert with the development of chronic inflammatory diseases [6,8]. The lungs and airway harbor a diverse composition of microbes and to date, many studies have confirmed that the microbiome of the respiratory tract not only can confer susceptibility to or cause respiratory diseases but can also be affected by the pathological activities of respiratory diseases and responses to treatment [9-11]. The presence of microbiota in the respiratory mucosa and the relationship between alterations in the constituents and respiratory immunity suggest that the airway microbiota play a role in chronic airway diseases, such as asthma, chronic obstructive pulmonary disease, and cystic fibrosis [12,13].
Inhaled allergens first encounter the host immune system in the nasal mucosa, and the microbial characteristics of the nasal mucus directly impact the mechanisms of initial allergic responses in the nasal epithelium [14]. Research on the role of the microbiome in sinonasal diseases is rapidly expanding, and insights into the microbiota and dysbiosis of the allergic nasal mucosa provide fundamental information regarding susceptibility to allergens and its relationship to allergic inflammation [15,16]. Further studies must be conducted to draw firm conclusions on potential functions or alterations of the airway microbiota in AR, but current evidence suggests that the microbiota in the airway exert an impact on disease progression and exacerbation. Therefore, alterations to the airway microbiota can influence the consequences of AR and investigating the microbial balance might provide new insights into the pathogenesis of these diseases and fundamental therapeutic strategies.
Based on the current data, we identified Ralstonia, Staphylococcus, and Propionibacterium species (spp.) as the most abundant constituents in healthy nasal mucus and contributors to significant dysbiosis of the microbial composition in the nasal mucus of patients with AR. Our study presents evidence that colonization of Staphylococcus spp. was altered most significantly in the nasal mucus of patients with AR, and Staphylococcus aureus contributes to dysbiosis in allergic nasal mucus in a manner dependent on patientsŌĆÖ clinical factors.
MATERIALS AND METHODS
Participant recruitment
A total of 104 participants (45 healthy participants, 59 AR patients) were enrolled in this study, and the difference of nasal microbial genus was first analyzed using the nasal mucus from 72 participants (30 healthy participants and 42 patients with AR), stratified by sex as 44 men (mean body mass index, 21.8 kg/m2) and 28 women (mean body mass index, 22.3 kg/m2) with a mean age of 32.5 years. Eligible individuals for inclusion in this study included those referred to the Department of Otolaryngology Chung-Ang University College of Medicine from November 2013 to April 2014. Intranasal endoscopy, computed tomography of the paranasal sinus, and AR tests were performed before intranasal sampling; none of the study participants showed signs of upper airway infection. Subjects who had taken any kind of antibiotics 2 months prior; were pregnant or smoker; had diseases and medication histories related to asthma; and/or had any other chronic diseases, such as atherosclerosis, hypertension, arrhythmia, congestive heart failure, diabetes mellitus, osteoporosis, hepatitis, cancer, and autoimmune or neurological diseases, were excluded.
The Institutional Review Board approved the protocol of this study (No. C2013158 [1118]). Study participation was voluntary, and written informed consent was obtained from all participants. The nasal microbial composition at the species level was further analyzed using nasal mucus from 32 participants, stratified as 15 healthy participants (mean age, 34.3 years) and 17 patients with AR (mean age, 37.1 years), who were referred to the Department of Otorhinolaryngology Seoul National University College of Medicine primarily for septal surgery between March 2018 and January 2019 (No. C2012248 [943]). To confirm AR, they underwent a skin prick test or a multiple allergen simultaneous test (MAST) using the Optigen allergen-specific IgE assay system Korean Inhalant Panel (Minaris Medical America, Mountain View, CA, USA) for the detection of allergens and specific IgEs. Among them, 45 subjects were classified into healthy subjects who were negative for all antigens and had no symptoms such as runny nose, nasal obstruction, itching sensation and sneezing. Fifty-nine who were confirmed positive for a specific antigen and showed symptoms of rhinitis were classified as AR subjects.
Mucus collection and DNA extraction
Mucus was collected from the middle turbinate individually using sterile 3M Quick swabs (3M Microbiology Products, St. Paul, MN, USA) from 72 participants (30 healthy participants and 42 patients with AR) using a rigid 0┬░ endoscope in an operating room. The cotton swabs were inserted into the nasal cavities of the participants without touching either the nostril or the anterior part of the inferior turbinate, which are lined with stratified squamous epithelium, and were gently rotated around the middle turbinate lined with respiratory epithelium. The swabs with mucus were fixed in a fixative solution and transported immediately to the laboratory for identification and microbial analysis. Each swab sample was centrifuged at 12,000 ├Śg for 15 minutes, and the supernatant was removed. To detect contamination, negative controls were prepared and subjected to the same procedures. The first step in the analysis was extraction of DNA from the bacterial pellet; here, bacterial DNA from middle turbinate mucosal swab samples was extracted with the Fast DNA Spin Kit for Soil (MP Biomedicals, Solon, OH, USA). The extracted DNA was dissolved in sterile water containing 40 ┬Ąg/mL RNase A and quantified with a Nano Quant Infinite M200 spectrophotometer (Tecan, M├żnnedorf, Switzerland) as the ratio of absorbance values at 260 and 280 nm (A260/A280).
Microbiome analysis
DNA from each swab sample was amplified in the hypervariable V1ŌĆōV3 region of the 16S ribosomal RNA (rRNA) gene before pyrosequencing. V1ŌĆōV3 amplification involved a specific primer set: 8F (5┬┤-CTGCTGCCTYCCGTA-3┬┤) as the forward primer and 530R (5┬┤-GTATTACCGCGGCTGCTG-3┬┤) as the reverse primer, with a 10-bp multiplex identifiers (MID) sequence. Polymerase chain reaction (PCR) was conducted on a T-professional Thermal Cycler (Biometra, G├Čttingen, Germany) under the following conditions: initial denaturation at 94┬░C for 5 minutes, followed by 35 cycles of denaturation at 94┬░C for 1 minute, annealing at 58┬░C for 1 minute, extension at 72┬░C for 1 minute, and a final extension at 72┬░C for 10 minutes. The PCR products were purified using the AccuPrep PCR Purification Kit (Bioneer, Daejeon, Korea) and analyzed by electrophoresis in a 1.2% agarose gel. Equal amounts of purified PCR products were pooled for 454 pyrosequencing, which was performed using a Roche 454 GS-FLX Titanium system (Roche Molecular Systems, Branchburg, NJ, USA). After a sequencing run was finished, the sequence and quality data were recovered as MID sequences. Quality-control efforts were conducted to eliminate trimmed sequences with ambiguous base calls, low-quality sequence ends, read lengths <200 bp, and quality scores <Q-25.
Sequence analysis
Bacterial composition was estimated from the data using the Quantitative Insights into Microbial Ecology (QIIME 1.9.1) pipeline [17]. Briefly, QIIME was used to assess phylogenetic and operational taxonomic units (OTUs). First, it was used to de-multiplex the barcoded reads and perform chimera filtering. Filtered sequence reads were grouped into OTUs at a sequence similarity level of 97%, which approximates species-level phylotypes. Next, the taxonomy of the OTUs was assigned, and sequences were aligned with Greengenes and PyNAST. The bacterial taxonomic abundance data were imported into R version 3.2.3 (R Foundation for Statistical Computing, Vienna, Austria) with the phyloseq package to analyze richness, diversity, and ordination plot [18]. Also, the lme4 package in R was used for fitting linear mixed-effects models (LMEMs) [19]. The compositional differences of nasal microbiome among healthy volunteers and AR patients were evaluated by non-parametric Wilcoxon test and the value has been adjusted by Holm-Bonferroni method for multiple comparisons. A P-value of <0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism (version 6.0; GraphPad Software, San Diego, CA, USA) to identify relative frequencies of differences in microbial genera. Data were considered significantly different at P<0.05.
RESULTS
Differences in microbial phyla and genera in the nasal mucus of healthy participants and patients with AR
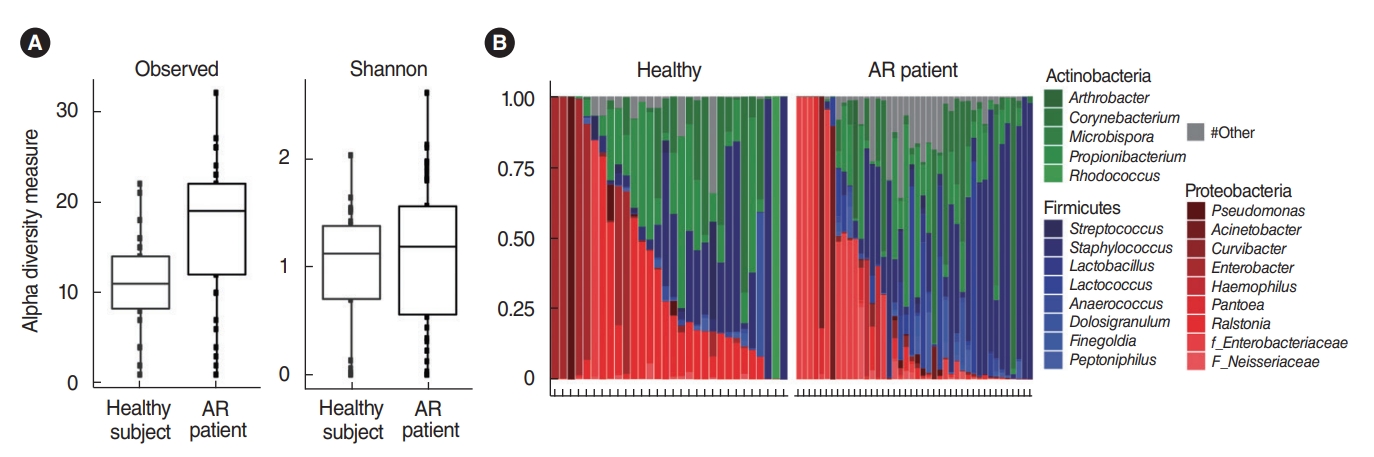
In total, 364,923 sequence reads of 16S rRNA genes derived from the nasal mucus swab samples of the middle turbinate were obtained from the genomic DNA samples of 42 patients with AR and 30 healthy participants. The raw data were submitted to the National Center for Biotechnology Information Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA236631. In terms of alpha-diversity, using the qualified sequence reads, although we found that typical healthy participants demonstrated greater genus-richness than patients with AR (Wilcoxon test, P<0.001), there were no significant differences in the Shannon index of the microbial community of the nasal mucus (Wilcoxon test, P=0.39) (Fig. 1A). This means that the occurrence of AR might not be explained by alpha dysbiosis in the microbiota of the nasal mucus.
The bacterial composition in the nasal mucus from both the healthy participants and the AR patients showed variable composition patterns. Taxa with an average abundance of at least 1% are shown, while those with less than 1% were reported as ŌĆ£other.ŌĆØ Although the number of genera to which symbiotic microorganisms belonged was higher in the AR patient group than in healthy participants, three phyla (Firmicutes, Proteobacteria, and Actinobacteria) were the most dominant in the nasal mucus of both groups. (Fig. 1B). Specifically, the Proteobacteria (42.34%┬▒34.4%) and Actinobacteria (33.73%┬▒28.0%) phyla were represented relatively well in the healthy participants, while the Firmicutes (41.23%┬▒33.6%) phylum was predominant in patients with AR based on LMEMs (Table 1).
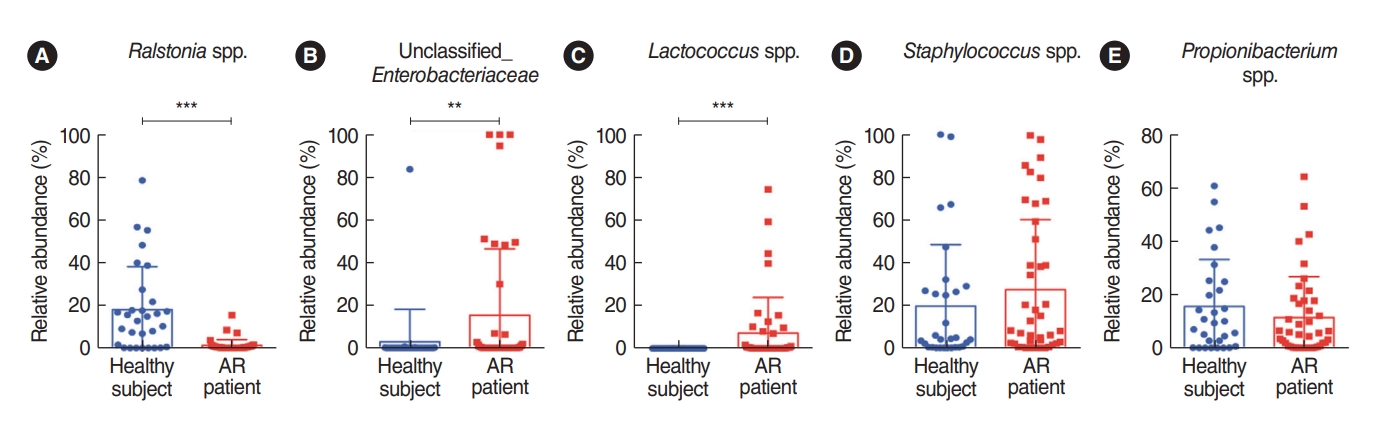
To characterize key genera of the microbiota found in the nasal mucus collected from healthy participants and patients with AR, the bacterial genera were compared using the non-parametric Wilcoxon test (Fig. 2). The results revealed that 11 nasal microbial genera of AR patients and healthy controls were significantly abundant. Of the identified 11 genera, three taxa were significantly different between the groups, and the Ralstonia genus was significantly more common in the nasal microbiome in healthy participants (Fig. 2A). In addition, both the unclassified Enterobacteriaceae and Lactococcus genera were more abundant in patients with AR (Fig. 2B and C). Although the abundance of these bacteria was not significantly different, the Staphylococcus genus was relatively more abundant in the nasal mucus of patients with AR and the rate of Propionibacterium genus was relatively higher in the nasal mucus of healthy participants (Fig. 2D and E). Despite the abundance of unclassified Enterobacteriaceae (Proteobacteria phylum), the Firmicutes phylum, including the Enterococcus and Staphylococcus genera, was more characteristic of the nasal mucus of AR patients. Based on these findings, we found that the composition of the symbiotic microbiome in the nasal mucus was distinctively different at both phylum and genus levels between healthy participants and AR patients. We performed an additional microbial analysis focusing on the genus level, which showed a distinctive distribution in the nasal mucus between healthy participants and AR patients.
Analysis of microbial genera showing differences in the composition of nasal mucus between healthy participants and patients with AR
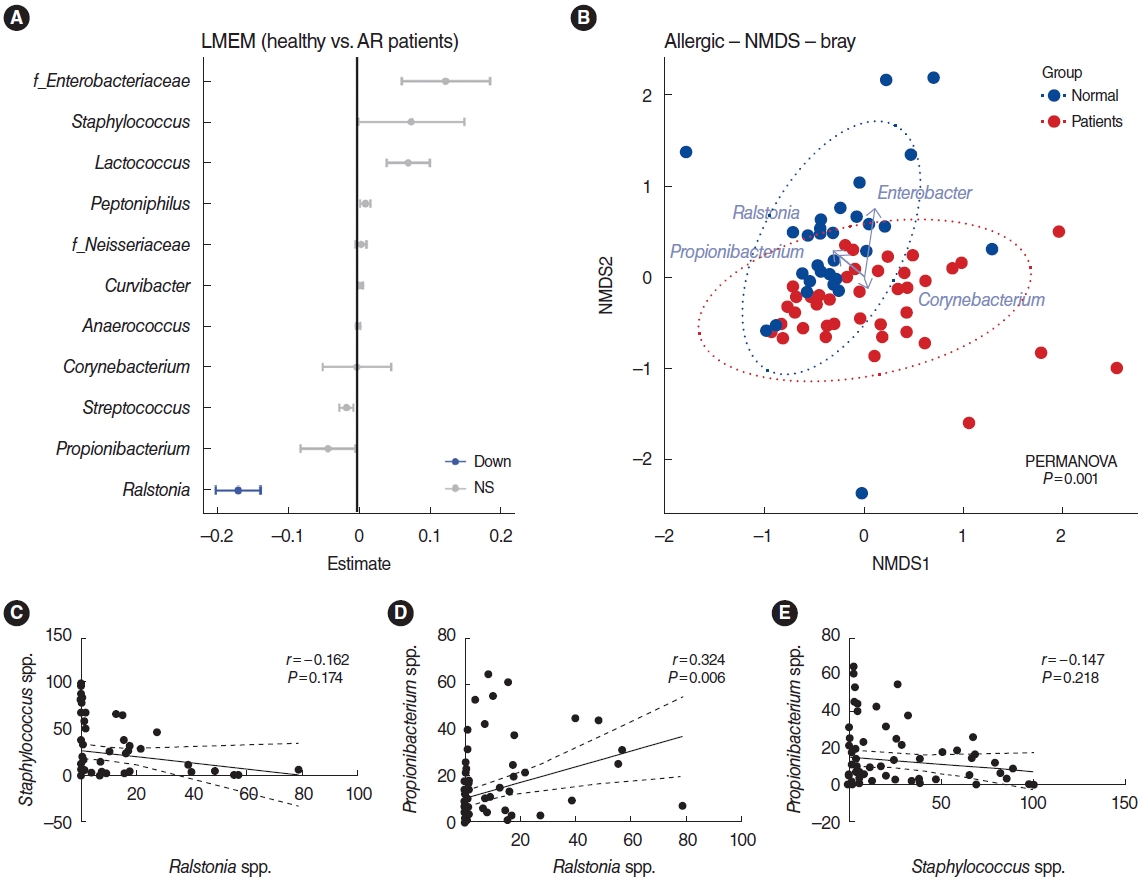
A comparison of the relative microbiome abundance between healthy participants and AR patients is shown based on the profiling of strains that were dominant or significant. The Staphylococcus and Propionibacterium genera did not show significant differences, but they appeared as the dominant strain within each group. In addition, although a difference between two groups was confirmed, there might have been no statistical significance due to the large standard error of the two groups. As the next step, a LMEM analysis was performed to elucidate differences in the distribution of key genera between the healthy participants and the AR patients. Eleven of the 22 genera (unclassified Enterobacteriaceae, Staphylococcus, Lactococcus, Peptoniphilus, unclassified Neisseriaceae, Curvibacter, Anaerococcus, Corynebacterium, Streptococcus, Propionibacterium, and Ralstonia), were identified in the LMEM results (Fig. 3A). Among those bacteria, only the Ralstonia genus was significantly over-represented in the healthy group (P<0.001). Whereas the Ralstonia genus was more abundant in the nasal mucus of healthy participants, the unclassified Enterobacteriaceae, and Lactococcus genera were more dominant in the nasal mucus of patients with AR.
We examined whether the two groups could be distinguished from each other. To this end, non-metric multidimensional scaling (NMDS) based on BrayŌĆōCurtis dissimilarities was used for analysis, and clustering of the nasal microbiome in both healthy participants and patients with AR was observed. Permutational multivariate analysis of variance confirmed that sample type was the dominant driver of variation (P=0.001). In addition, biplot vectors showed the four genera with the strongest contribution to sample dissimilarity. The NMDS biplot used an extension of the vegan libraryŌĆÖs bioenv function to find the best set of bacterial taxa with community dissimilarities (Bray) based on Pearson correlations and then plotted them as vectors. As expected, the Ralstonia genus of the Proteobacteria phylum was found in higher proportions in the nasal mucus of healthy participants, in concert with abundance of both the Propionibacterium genus of the Actinobacteria phylum and the Enterobacter genus of the Proteobacteria phylum, whereas the nasal mucus of patients with AR was predominated by the Staphylococcus genus of the Firmicutes phylum with the co-presence of a minor bacterium (the Corynebacterium genus), whereas the nasal mucus of AR patients was predominated by the Corynebacterium genus of the Actinobacteria phylum (Fig. 3B).
To confirm the patterns in terms of relative abundance of bacterial taxa, Spearman correlation analysis was conducted (Fig. 3C-E). The data showed that AR patients with a high distribution of the Ralstonia genus had a relatively low distribution of the Staphylococcus genus (Spearman rho=ŌłÆ0.1621, P=0.174) and AR patients with a high distribution of the Propionibacterium genus also had a lower distribution of the Staphylococcus genus (Spearman rho=ŌłÆ0.1471, P=0.218). As expected, the distribution of the Ralstonia and Propionibacterium genera showed a positive correlation (Spearman rho=0.32, P=0.006). These findings demonstrate that the Firmicutes phylum was most abundant in the nasal mucosa of patients with AR, and the distribution of the Staphylococcus genus was altered substantially in the nasal mucus of patients with AR. Although statistical significance was not shown in all analyses, the abundance of the Staphylococcus genus in the nasal mucosa of AR patients showed a negative correlation with the abundance of the Ralstonia and Propionibacterium genera.
Dysbiosis of nasal microbial species in AR associated with patientsŌĆÖ clinical characteristics
As a next step, we analyzed alterations in the microbial composition in the nasal mucus of patients with AR at the bacterial species level. To further assess alterations in microbial composition at the levels of bacterial phyla and species, the microbial compositions of the middle turbinate mucus in healthy participants (n=15) and patients with AR (n=17) were evaluated. We re-confirmed the results, finding the same trends as shown in Fig. 1B, and the Proteobacteria (30.2%), Firmicutes (14.4%), Actinobacteria (14.2%), and Bacteroidetes (7.1%) phyla predominated in the nasal mucus of healthy participants. Further, pyrosequencing data revealed that the proportions of the Proteobacteria, Actinobacteria, and Bacteroidetes phyla were reduced in the nasal mucus of patients with AR, and the proportion of the Firmicutes phylum was elevated (Fig. 4A). Based on a sequence identity of at least 97%, Nocardia coeliaca (20.01%, Actinobacteria phylum), Staphylococcus epidermidis (14.45%, Firmicutes phylum), Corynebacterium accolens (7.15%, Actinobacteria phylum), and Citrobacter koseri (6.77%, Proteobacteria phylum) were the most abundant microbial species in the nasal mucus of healthy participants. In particular, a greater abundance of S. epidermidis, C. accolens, and N. coeliaca, accounting for 41.55% of mapped sequences in the nasal mucus of healthy participants. In contrast, the distribution of microbial species was altered greatly in the nasal mucus of patients with AR, where S. aureus demonstrated the greatest abundance of mapped sequences and the largest expression in terms of the microbial distribution (37.69% in AR vs. 0.21% in healthy participants) (P<0.001) (Fig. 4B). C. accolens and N. coeliaca were distributed at relatively high proportions (P=0.012), but the composition of S. epidermidis was significantly reduced in the nasal mucus of patients with AR (3.24%) (P=0.004). The proportions of S. epidermidis and S aureus differed significantly among patients with AR, with a maximum of 96.92%, and the lowest abundance of Staphylococcus genus in the nasal mucus of patients with AR was 0.94% (Fig. 4C).
We explored whether alterations of Staphylococcus spp. in the nasal mucus of patients with AR might be correlated with the clinical characteristics of AR patients. In eight of 17 patients with AR, S. aureus was the most abundant species in the microbiome of the nasal mucus, and more than 40% of nasal symbiont microorganisms were identified as S. aureus (Fig. 4C). Although no significant correlation was found between the proportion of S. aureus and other clinical factors, both the age and height of patients with AR showed statistically significant correlations with the abundance of S. aureus in the nasal mucus. Interestingly, 11 patients with AR were confirmed to have strongly positive (>3+) results for Dermatophagoides pteronyssinus and Dermatophagoides farina based on allergy tests, such as an allergic skin test or MAST (Table 2). The rates of positive and negative reactions to house dust mites (HDMs) showed a statistically significant correlation with a higher abundance of S. aureus in these patients (Fig. 4D, Table 3). Considering these findings, we provide a note of caution that S. aureus-derived dysbiosis was distinctive in the nasal mucus of patients with AR, and this dysbiosis might be correlated with patientsŌĆÖ age, height, and antigen-positivity for HDM.
DISCUSSION
In the present study, we showed a significantly distinctive colonization pattern of the Firmicutes, Actinobacteria, and Proteobacteria phyla in the nasal mucus of patients with AR compared to healthy participants. In addition, significantly different distributions of the Ralstonia, Enterobacteriaceae, and Lactococcus genera were observed in the nasal mucus of healthy participants and patients with AR. Our findings clarify that symbiotic dysbiosis was more pronounced in the nasal mucus of patients with AR. The Staphylococcus genus exhibited the most noticeable alterations in composition between healthy participants and patients with AR, and the composition of S. aureus was most abundant in the nasal mucus of AR patients depending on their age, height, and antigen-positivity for HDM.
The nasal epithelium is the first target organ for environmental allergens, and recent work has highlighted its critical role as a barrier that restricts host exposure to allergens in AR [3,4]. The microbiome colonizes the nasal mucus, where contact with inhaled pathogens or allergens occurs, and might engage in functional crosstalk with the nasal epithelium, especially concerning the regulation of immune mechanisms in the upper airway [20]. Therefore, understanding compositional changes in the microbiome and identifying dominant microbial species in the nasal mucus might be essential for understanding the definite pathophysiologic mechanism of upper airway diseases, including AR. In recent years, as the importance of microorganisms has been emphasized, analyses of microbial profiles or alterations between health and disease conditions have become accessible through the development of culture-independent technologies such as next-generation sequencing, and research on the distribution of symbiotic microorganisms has become possible up to the species level [21].
In particular, the importance of the nasal microbiome, especially with respect to Th2 cytokine-regulated immune responses, has been increasingly recognized [22]. Therefore, we extended this paradigm to the nasal microbiome in the field of AR and noted marked alterations in microbial composition at the species level in the nasal mucus of patients with AR. To obtain more information about the microbial symbionts in the nasal mucus in this study, mucus samples were obtained via swabs of the middle turbinate. Nasal mucus, which is secreted by epithelial cells, was analyzed to determine the composition of the nasal microbiome community that resides in the nasal mucosa. We think that the mucosa around the middle turbinate is correlated highly with the microbial environment as relevant for practical purposes, with little influence of the external environment, and it is a clinically appropriate source of samples to analyze changes in the distribution of symbiotic microorganisms in nasal diseases such as AR.
Out data showed that the Proteobacteria, Actinobacteria, and Firmicutes phyla were relatively more abundant in the nasal mucus of healthy participants. In healthy participants, there was a significant abundance of the Ralstonia, Staphylococcus, and Propionibacterium genera, and N. coeliaca and S. epidermidis were the most dominant microbial species. Our previous study showed that the Staphylococcus (Firmicutes phylum) and Corynebacterium (Actinobacteria phylum) genera were the most abundant commensal organisms and S. epidermidis was the most common nasal symbiont of the identified bacterial species in human nasal mucus [23,24]. The current findings revealed that the microbial composition was altered significantly in the nasal mucus of patients with AR. Interestingly, the proportions of the Proteobacterium and Actinobacteria phyla, as symbiotic microbiota, were reduced in the nasal mucus of patients with AR, while the proportion of Firmicutes phylum was elevated. This difference also was observed in microbial genera in the nasal mucus of patients with AR. The proportions of the Ralstonia and Propionibacterium genera were lower, and the Staphylococcus genus was more dominant in the nasal mucus of patients with AR. In addition, the proportions of the Ralstonia and Propionibacterium genera were correlated inversely with that of the Staphylococcus genus in each patient with AR.
Ultimately, the nasal mucus of AR patients induces a biological environment where certain commensal microorganisms can better exist, and more research is needed to analyze the relationship between dysbiosis of the nasal microbiome at the species level and the pathophysiology of AR. In the nasal microbiome of patients with AR, S. aureus colonization was significantly predominant, in concert with a high abundance of Staphylococcus spp. Our previous study showed that the distribution of S. aureus was significantly elevated in the nasal mucus of patients with AR, to the point that it constituted more than 20% of the identified nasal microbiome, and S. aureus colonization was most highly abundant relative to the composition of the normal mucus [25]. Thus, we sought to understand the contributions of Staphylococcus spp. to the pathogenic mechanism of AR as nasal symbionts and the synergism of Staphylococcus spp. colonization with Th2-related allergic inflammation in the nasal mucosa. Both S. epidermidis and S. aureus are well-known human pathogens that cause serious opportunistic infections of the respiratory tract [26,27]. However, Staphylococcus spp. are detected commonly in the normal microflora of the skin, intestine, and upper airway, and many studies have identified a possible role of the Staphylococcus genus as symbiotic commensal organisms involved in allergic diseases [28-31]. Patients with allergic eczema were more likely than healthy participants to have skin colonized with S. aureus, and disease severity was associated with the degree of S. aureus colonization [29]. The dominant distribution of S. aureus in the upper respiratory tract of patients with asthma and aspirin sensitivity has been reported, with proportions of up to 87.5%, and greater nasal S. aureus colonization was found to be significantly related to asthma [30]. In addition, patients with AR were colonized more frequently with nasal S. aureus or sensitized to S. aureus enterotoxins than healthy participants [32]. These findings support the involvement of S. aureus in allergic immune responses and the correlation of S. aureus colonization with positive or negative regulation of Th2 inflammation. Abnormalities in the human microbiota are associated with the etiology of AR, and research has sought to characterize the microbiota of the site of AR (i.e., the inferior turbinate) in subjects with AR and healthy controls and to examine the relationship of the mucosal microbiota with disease occurrence, sensitized allergen number, and allergen-specific and total IgE levels. Microbial dysbiosis correlated significantly with total IgE levels, representing the combined allergic response, but not with disease occurrence, the number of sensitized allergens, or HDM allergen-specific IgE levels [33].
Our data also indicate that the nasal mucosa in AR patients might provide a more suitable environment for the nasal symbiont S. aureus to survive, resulting in its greatest abundance in the microbiome in the nasal mucus of patients with AR. Intriguingly, the proportion of S. aureus in the nasal mucus of patients with AR was positively correlated with a positive reaction for HDM. We did not determine how nasal Staphylococcus microbiome colonization became more abundant in the nasal mucus of patients with AR and whether this played a critical role in regulating the Th2 immune response in patients with AR. Instead, we only surmised that the human nasal mucosa could become a more favorable environment for the survival of S. aureus depending on AR patientsŌĆÖ clinical factors, such as age or height. In addition, S. aureus colonization was more abundant when AR patients were sensitized to HDM, and we presume that the metabolites secreted by HDM after exposure to the nasal mucus and the proteolytic enzymes of HDM might induce alterations in the proteomic composition of the nasal mucus.
This study evaluated the alterations in the nasal microbiome that occurred in the nasal mucus of patients with AR and documented significant proportional changes in the nasal mucus at the levels of microbial genera and species. Our study highlights S. aureus-dominant dysbiosis of the nasal mucus of patients with AR and enhances our understanding of microbial alterations related to clinical characteristics of patients with AR, including those related to specific allergens. Future research directions need to include manipulation of the dominant nasal microbiome for therapeutic advances in managing AR and might provide a basis for research on biologics for AR treatment.
HIGHLIGHTS
Ō¢¬ A significantly distinctive colonization pattern of the Firmicutes, Actinobacteria, and Proteobacteria phyla was observed in the nasal mucus of patients with allergic rhinitis (AR) compared to healthy participants.
Ō¢¬ Nasal commensals in both the unclassified Enterobacteriaceae and Lactococcus genera were more abundant in the nasal mucus of AR patients.
Ō¢¬ The distribution of the Staphylococcus genus also exhibited noticeable alterations between healthy participants and patients with AR.
Ō¢¬ Dysbiosis of Staphylococcus aureus was present in the nasal mucus of AR patients, and the abundance of S. aureus in AR patientsŌĆÖ nasal mucus depended on their age, height, and sensitization to house dust mites.