INTRODUCTION
Studies on the pathogenesis of chronic rhinosinusitis (CRS) are challenging, as they require longitudinal cohorts and analyses prior to the onset of disease. Although significant findings have been made regarding the pathophysiology of CRS, the majority of these studies have been retrospective or cross-sectional. However, many CRS patients subjectively recall that their symptoms began with an upper respiratory infection (URI) that progressively became more severe and chronic in nature. URIs are common viral infections affecting the nose, throat, and airways, and can last between 7 and 11 days. In some patients, a URI can progress into acute rhinosinusitis (ARS). ARS features an increase in symptom severity for more than 10 days and is frequently associated with facial pain/tenderness, hyposmia/anosmia, nasal obstruction, and mucopurulent drainage. In certain cases, these symptoms persist for at least 12 consecutive weeks and meet the criteria of CRS (Table 1) [1]. CRS is a complex inflammatory disorder that affects between 2% and 16% of adults in the United States, with estimated healthcare costs between 4 to 12 million USD [2,3].
Viruses are a frequent etiologic factor for URIs, are frequently identified in the sinuses of patients with CRS, and trigger CRS exacerbations [4]. Therefore, investigating the role of viruses may provide insights into the pathogenesis of CRS. In this review article, we will discuss the role of viruses and their associated immunologic responses and contributions to inflammation in CRS. This information may allow us to target pathways early in the pathogenesis of CRS, thereby playing a significant role in slowing the progression of this chronic disease.
WHAT VIRUSES ARE SEEN IN CRS?
Several cross-sectional studies have identified the types of viruses associated with CRS. Cho et al. [4] found that CRS patients had higher proportions of respiratory viruses in their nasal secretions than the control group of patients without CRS. Of the viruses identified, rhinovirus (RV) infection in lavage and mucosal samples was significantly associated with CRS patients compared to controls. In the same study by Cho et al. [4], parainfluenza virus (PIV) , influenza virus, and respiratory syncytial virus (RSV) were all also found in the nasal lavage samples of patients with CRS. However, only RV and PIV were detected at higher rates among CRS patients than in the control group. In another study by Ramadan et al. [8], polymerase chain reaction (PCR) confirmed that 20% of the CRS patients’ samples were positive for RSV RNA, but none were positive for adenoviral DNA. In a third study by Abshirini et al. [9], reverse-transcription PCR showed that 28.94% of their sample of patients with CRS had RV and 11.84% had RSV. One of the most significant pathogens in terms of CRS is human rhinovirus (HRV). HRVs frequently cause the common cold, as well as in CRS. There are three main subgroups of HRV: HRV-A, HRV-B, and HRV-C [10]. However, the majority of studies did not identify HRV species. Willis et al. [11] reported that HRV-C infections were associated with more sever sinus symptoms, which is similar to findings seen in asthma.
In summary, the viruses that are frequently associated with CRS are RV, RSV, PIV, and influenza virus. Other viruses, such as adenoviruses, did not show a strong correlation with CRS populations compared to controls. The coronavirus disease 2019 (COVID-19) pandemic has also brought an increased focus on the role of viruses in sinonasal disease. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the etiologic virus responsible for COVID-19, and its receptor, angiotensin-converting enzyme 2 (ACE2), is highly expressed in the nasal and sinus epithelia [12]. RNA viruses and respiratory diseases can increase ACE2 expression. RV and its subtypes RV-A and RV-C significantly upregulate ACE2 expression, including the truncated isoform delta-ACE2, in human nasal airway epithelial cells [13,14]. Similar symptoms, such as olfactory dysfunction, are shared by COVID-19 and CRS, but no conclusive results have been reported regarding whether patients with SARS-CoV-2 have an increased risk of CRS [15,16].
VIRUS RECEPTORS AND TARGETS IN THE UPPER AIRWAY
Several viruses are known to be associated with CRS, and it is important to analyze and study their mechanisms of infection, as well as the specific receptors each virus targets. These RNA viruses include RV, influenza virus, RSV, and PIV (Table 2) [17]. RV and its subtypes invade the host cells using three types of cellular membrane glycoproteins: intercellular adhesion molecule 1 (ICAM-1), low-density lipoprotein receptor (LDLR) family members, and cadherin-related family member 3 (CDHR3) [18-21]. The ICAM-1 receptor is located in the plasma membrane and cytoplasm on the apicolateral portions of the airway epithelial cells. ICAM-1 mediates leukocyte adhesion and regulates endothelial cell shape, as well as blood vessel barrier function [22]. When expressed by dendritic or natural killer cells, ICAM-1 plays a significant immunological role in T-lymphocyte binding and the formation of immune synapses. A more recently discovered role of ICAM-1 is promoting macrophage efferocytosis, or the removal of dying cells, which is important for resolving inflammation and tissue homeostasis [22,23]. In the presence of inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and interferon (IFN)-γ, ICAM-1 expression increases, while glucocorticoids inhibit its expression. ICAM-1 activates transcription factors, increases cytokine production, and is used by a majority of RV-A and all of RV-B subgroups [18,37,38]. The LDLR family members comprise a group of endocytic cell surface receptors that bind to extracellular ligands (e.g., lipoproteins, exotoxins, and lipid-carrier complexes) and bring them into the cell. These receptors mediate lipoprotein ligands including chylomicrons, low-density lipoprotein, intermediate-density lipoprotein, or very low-density lipoprotein. LDLR proteins normally play a significant role in cardiovascular disease and lipoprotein homeostasis, as well as atherosclerosis [39]. They are located in recycling endosomes, or less commonly, the plasma membrane, and they target 12 known RV-A types [18,39]. Cadherins are a group of transmembrane glycoproteins whose functions include adhesion, cell signaling, and mechanical transduction. CDHR3 receptors are highly expressed in the airway epithelium and are located in the plasma membrane. The CDHR3 receptor, which has been found to be strongly associated with asthma exacerbation in children, mediates virus binding and replication for the subgroup RV-C [18,21,40].
Influenza virus contains a viral attachment protein called hemagglutinin (HA), which is a naturally occurring glycoprotein causing agglutination of red blood cells. HA in influenza virus binds to and utilizes sialic acid-containing molecules as receptors to gain entry into the cell. This leads to infection of multiple cell types utilizing these abundant molecules as receptors, resulting in viral binding to nonproductive sialic acid-containing molecules. Therefore, influenza virus also contains a second viral surface protein, neuraminidase, that can cleave sialic acid to release the virus after binding to any molecules that do not lead to viral infection. Influenza virus primarily targets airway epithelial cells using α2,6-type receptors in humans [27].
RSV targets ciliated epithelial cells in the airways in which the RSV-fusion (RSV-F) glycoprotein binds to the cellular receptor human nucleolin. However, it has been suggested that RSV also uses signaling receptors that activate kinases and mediate its entry. Griffiths et al. [30] found that the insulin-like growth factor-1 receptor (IGF1R) inhibitor PQ401 and a polyclonal anti-IGF1R antibody reduced infection by equivalent amounts, and that insulin-like growth factor (IGF)-1 significantly enhanced RSV infection. There was also colocalization of IGF1R with RSV particles in cells, suggesting that RSV may interact with IGF1R during virus entry. Anderson et al. [31] found that the human chemokine receptor, C-X3-C motif chemokine receptor 1 (CX3CR1), may be a receptor for RSV infection, as RSV viral loads were greatest in cells that expressed CX3CR1. Meanwhile, blocking the interaction resulted in reduced RSV viral loads.
Human PIVs have three types of receptors: high-power field (HPF) 1, 2, and 3, where each type targets different areas of the respiratory tract. HPF3 targets the upper respiratory tract, leading to respiratory diseases like bronchiolitis and pneumonia. Similar to influenza virus, the receptor-binding HA-neuraminidase interacts with sialic acid-containing molecules on the cell surface for HPF3-mediated membrane fusion, as well as using the HPF3 fusion protein [35].
IMMUNOLOGIC RESPONSES TO VIRUSES
There are two main immune responses to viral infection. Type 1 immune responses (Th1) are characterized by the production of cytokines that exhibit pro-inflammatory responses, such as IFNs. Type 2 immune responses (Th2) are characterized by eosinophilic and immunoglobulin E responses as well as ILs (i.e., IL-10, IL-4, IL-13, and IL-5). These responses are associated with atopy and are anti-inflammatory. In a healthy immune system, Th1 and Th2 responses balance each other, leading to an optimal immune response [41]. However, a dysregulated immune response to viral infections can result in the activation of airway remodeling, the epithelial-mesenchymal transition, and epithelial barrier breakdown, which are central to the pathogenesis of CRS [42].
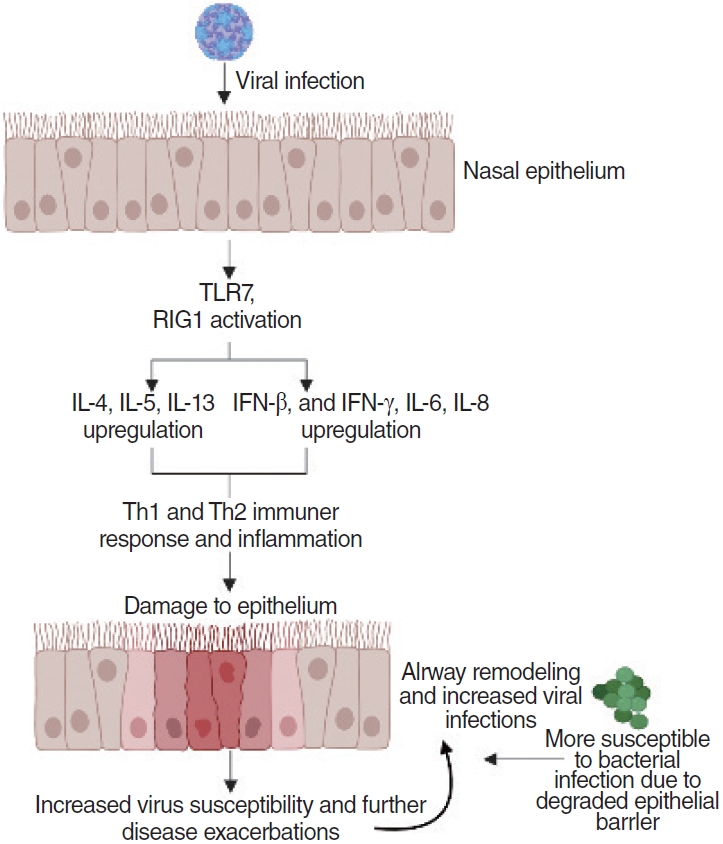
RV infection occurs at the airway epithelium and activates Toll-like receptor 7 (TLR7) and retinoic acid-inducible gene I (RIG-1), triggering the induction of cytokine expression (type I and type III IFNs). IFNs are classified based on their amino acid sequence and structure. Type I IFNs bind and signal through the IFN-α and beta receptor subunit (IFNAR)-1 and IFNAR2 receptor complex, while type III IFNs signal through IFNλR1 and IL-10R2 receptors [43-45]. Both type I and type III IFNs use the same Janus kinases that initiate IFN-mediated signaling cascades for signal transduction, but structurally, type I IFNs have longer and straighter α-helices than type III IFNs. and type III IFNs closely resemble the structure of IL-22 from the IL-10 family of cytokines [46-50]. Using human nasal epithelial cells, Tan et al. discovered that RV infection induced the expression of CXCL-9, CXCL-11, IFN-γ-induced protein 10 (IP-10), and regulated upon activation, normal T cell expressed and presumably secreted (RANTES), which are all components of the type 1 immune response. Although cytokine and chemokine expression were dominated by the type 1 immune response, moderate expression of type 2 immunity genes was also discovered [25]. Kim et al. [51] found that the same cytokines were induced by RV in patients with and without CRS. However, there was a slight impairment of IFN-β protein production and a delay of melanoma differentiation-associated protein 5 mRNA expression. Other studies have found that RV-B releases fewer proinflammatory cytokines, chemokines, and IFNs than the other RV subtypes (RV-A and RV-C) [52]. Yeo and Jang [26] found that RV infection resulted in significantly decreased mRNA levels of tight junction components, such as zonula occludens-1, occludin, and claudin-1, as well as adherens junction components (e.g., cadherin-1) in epithelial cells (Table 2). Epithelial cells act as a barrier and the first line of defense against infections. Therefore, the loss of barrier function via degraded tight junction and adherens junction components can increase the risk of chronic infection because microbes and antigens are more likely to pass through a defective barrier [53].
RSV infections are one of the most common causes of respiratory infections in children and infants. Most studies detailing the immune response due to RSV used patients who had lower respiratory tract infections. In patients infected with RSV, IFN-γ levels were shown to be increased in both the nasal mucosa and the lungs. Additionally, patients with lower IFN-γ production had higher severity scores [29,54-56]. In terms of the Th2 response, increased levels of IL-4, IL-6, IL-9, IL-10, and IL-13 were found in the nasal washes of RSV-infected children [29,57,58]. According to data from one study, a predominance of Th2 cytokines over Th1 cytokines was associated with children with hypoxic RSV lower respiratory tract infections, suggesting that a Th2-biased response is associated with severe manifestations of RSV infection [29]. Interestingly, several studies on RSV have shown that its pathogenesis is dependent on age. Using mice models, Hijano et al. [32] found that type I IFNs, such as IFN-α, were differentially expressed based on age. Conversely, IL-33, a Th2-oriented cytokine, was released in large amounts following RSV infections in neonatal mice, but the response decreased in adult mice. A study by Saravia et al. [33] confirmed these results, finding that neonatal mice that were induced with RSV responded with high levels of IL-33 expression and significant increases in type 2 innate lymphoid cells, while adult mice failed to show either response. This study also found that among infants hospitalized with RSV infections, IL-33 and IL-13 levels were elevated (Table 2) [32].
PIVs primarily affect children and are associated with the induction of wheezing early in life. Yoshizumi et al. [34] determined that cells infected with PIV released greater amounts of IL-1β, IL-6, TNF-α, IL-1ra, IFN-γ, IL-2, IL-4, IL-5, IL-10, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, IL-8, IP-10, eotaxin, RANTES, platelet-derived growth factor BB, and vascular endothelial growth factor than cells with no PIV infection (Table 2).
Studies have shown that patients infected with influenza A virus have increased levels of IL-6, IL-8, TNF-α, IL-10, and IFN-γ in their nasal lavage samples (Table 2). These cytokines also correlate with disease severity—as levels increased, disease severity increased [28]. According to a study by Skoner et al. [59], IL-6 was determined to play a potential role in initiating symptoms of influenza A infection, while IL-8 did not.
As these upper respiratory viruses infect the epithelium, they trigger immune responses that induce the release of cytokines and chemokines via intracellular sensors (TLR7 and retinoic acid-inducible gene I). Cytokines and chemokines, such as IL-6 or IFN-γ, are induced and secreted by the intracellular sensors before recruiting neutrophils and macrophages that activate the Th1 immune response [28,34,43-45]. The immune response leads to inflammation in the infected areas, which coupled with the damage from the viral infection itself and from the viral elimination by lymphocytes (e.g., Th1 cells, cytotoxic T cells), results in damage to the epithelium [60]. Continuous viral infection and inflammation cause airway remodeling of the nasal epithelium and degradation of tight junctions and adherens junctions [26,53]. Disrupted mechanical barriers and deficiencies in both the innate and acquired immune system make the sinonasal mucosa more susceptible to antigenic exposition and stimulation, leading to either side of the spectrum of chronic inflammation. This results in increased viral susceptibility of the epithelium, allowing further disease exacerbations and greater potential for bacterial infections to occur (Fig. 1). As the epithelium becomes degraded, the persistent infections and immune responses lead to CRS and CRS exacerbations [62]. Epithelial damage has been observed in CRS with nasal polyps, and genetic deficiencies or environmentally induced damage of epithelial repair mechanisms may be associated with both forms of CRS [61,63,64].
RISK FACTORS FOR VIRAL INFECTIONS IN CRS
Several risk factors can facilitate CRS infections by contributing to viral binding, entry, replication, and the immune response. Epithelial barriers are critical in preventing viral binding and entry into sinonasal epithelia. Mutations in CDHR3, the primary viral receptor for RV-C, have been associated with an increased risk for CRS and asthma [40]. One hypothesis is that the rs6963770 single-nucleotide polymorphism may result in increased RV-C binding and modulate a dysregulated immune response [65]. Age is considered a risk factor for RSV infection, as high rates of serious RSV infections and hospitalizations are observed among infants. Additionally, the presence of underlying conditions such as prematurity, congenital heart disease, immunosuppression, and cystic fibrosis all increase the risk of developing severe RSV infections [66]. Similar to RSV, PIV infections are more common and tend to be more severe in infants and young children, or elderly with compromised immune systems [67]. For influenza, a study of children in Ontario, Canada found that asthma, regardless of the severity, was a significant risk factor associated with severe disease [68].
Allergic rhinitis and asthma have a strong tendency to co-occur with CRS, suggesting a common mechanism of disease. In all three of these type 2-mediated airway disorders, the epithelial barrier is compromised. This leakiness in the epithelial barrier is hypothesized to allow enhanced viral entry through the epithelia to trigger alarmin signals including thymic stromal lymphopoietin, IL-33, and IL-13, which can trigger type 2 activation of mast cells and eosinophils. Similar mechanisms of epithelial barrier dysfunction can be seen in prolonged exposure to tobacco and air pollution, which are highly associated with CRS risk [69-72].
Aside from risk factors pertaining to an increase in CRS risk, there are risk factors for acute CRS exacerbations that increase nasal and sinus symptom severity. General health risk factors include smoking, a higher body-mass index, previous sinus surgery, and a longer CRS status, while several seasonal components such as hay fever or the winter season also increase the risk of acute CRS exacerbation [58]. Comorbid predisposing factors include asthma symptoms, impaired mucociliary clearance, and atrophic rhinitis [73-75].
CRS-RELATED VIRUSES IN CHILDREN VERSUS ADULTS
Age is a strong risk factor for CRS. Children have 3–8 viral URIs per year compared to adults who only have 2–4 URIs [76]. Male children under the age of 3 more commonly contract respiratory illnesses than female children of a similar age, while the opposite is true as their ages progress [77,78]. Comorbid conditions such as allergic rhinitis were found in 36%–60% of pediatric patients with CRS [79-81]. Khoo et al. [82] found that asthma and wheezing exacerbations in children were more prevalent at younger ages. RV-C was the most frequently identified viral pathogen in these children, and several viruses including RSV, PIV, and influenza virus were also detected.
Certain viruses are more predominant in certain age groups. Interestingly, the studies examined in this review revealed that RSV and PIV are notably more prevalent in certain age groups. For example, children and the elderly are well documented as being more susceptible to RSV infection than adolescents or adults. Immature immune systems or low lymphocyte counts in infants and young children, as well as low levels of RSV-neutralizing antibodies in patients over 65, are factors that cause these age groups to be more susceptible to infection [83,84]. Furthermore, RSV pathogenesis differs in children and adults. For example, most adults and elderly people infected with RSV show symptoms similar to influenza infection, while infants and young children with RSV infections often progress to lower respiratory tract infections and wheezing [85-87]. Additionally, PIV infection often causes URIs in most healthy young adults, but more frequently leads to severe symptoms and lower respiratory illnesses in young children. Similarly to RSV, PIV infection is one of the leading causes of acute respiratory tract infections in young children under the age of 5, accounting for approximately 17% of hospitalizations [67,88,89].
CONCLUSION
In summary, CRS affects millions of people worldwide and poses a significant financial burden. Therefore, understanding the mechanisms of infection that drive its pathology is important. In order to devise effective therapies for patients with CRS, understanding the viruses, their mechanisms of infections, and their immune responses is crucial. RVs are frequently isolated in patients with CRS. RSV, PIV, and influenza virus are also isolated in patients with CRS. These four viruses have many similarities such as targeting epithelial airway cells and being RNA viruses. However, the prevalence, receptor type, and immune response vary from virus to virus.
RVs are the most widely and thoroughly studied viruses in terms of CRS specifically, and even in terms of URI and acute sinusitis. In the future, we hope to see more studies that detail the immune response of upper respiratory tract infections and CRS due to RSV, PIV, and influenza virus. Additionally, we hope to see more longitudinal studies that follow infants and young children infected with serious respiratory viruses and how those infections can contribute to the onset of more serious cases of URI, such as CRS, later in adulthood. Throughout this review, we also noticed a scarcity of papers on pediatric CRS. Research indicates that this time point is critical in understanding the development and onset of adult CRS. Thus, further studies are necessary to be better able to target and create new therapies.
HIGHLIGHTS
▪ The four most commonly isolated viruses in patients with chronic rhinosinusitis are rhinovirus, parainfluenza virus, influenza virus, and respiratory syncytial virus.
▪ Viral infection in the upper airways has been shown to degrade epithelial barrier function, and rhinovirus infection has been specifically shown to degrade tight junction and adherens junction components.
▪ Age is strongly associated with chronic rhinosinusitis risk.
▪ Viral infections linked with chronic rhinosinusitis are more prevalent in infants and children than in adults.