Research Progress on Non-coding RNAs in Cholesteatoma of the Middle Ear
Article information

Abstract
Cholesteatoma of the middle ear is a common disease in otolaryngology that is receiving increasing attention. It is estimated that over five million people around the world have suffered from middle ear cholesteatoma. The annual incidence of middle ear cholesteatoma has been reported to be 9.2 per 100,000 in adults and 3 per 100,000 in children. Without timely discovery and intervention, cholesteatomas can become perilously large and damage intratemporal structures, causing various intracranial and extracranial complications. No practical nonsurgical treatments are currently available. Although multiple hypotheses exist, research directions have consistently focused on cell proliferation, apoptosis, and bone destruction. Non-coding RNAs (ncRNAs), especially microRNAs (miRNAs), long ncRNAs (lncRNAs), and circular RNAs (circRNAs), have recently received increasing attention because of their key roles in gene expression, cell cycle regulation, and the development of many diseases. Although ncRNAs are not involved in protein translation, they are abundant in the genome, with only approximately 2% of genes encoding proteins and the remaining approximately 98% encoding ncRNAs. The purpose of this review is to summarize the current state of knowledge regarding the specific role of ncRNAs in middle ear cholesteatoma.
INTRODUCTION
Cholesteatoma of the middle ear is a common disease in otolaryngology that is receiving increasing attention due to its ability to cause recurrent ear discharge, deafness, vertigo, and even intracranial and extracranial complications such as facial palsy, septic meningitis, and brain abscess. Pathologically, middle ear cholesteatoma is defined as a cystic structure located within the tympanic papillae that, although not a true cancer, can gradually expand as the keratinized epithelium in the cystic pouch continues to shed and accumulate, which involves the destruction of adjacent tissue structures. Cholesteatoma is classified as congenital or acquired and acquired cholesteatoma can be further classified as primary or secondary. The mechanism of acquired cholesteatoma development remains unclear, but there are four primary hypotheses [1,2]: (1) the squamous metaplasia theory, (2) the basal cell proliferation theory, (3) the epithelial migration theory, and (4) the retraction pocket theory. Although multiple hypotheses exist, research directions have consistently focused on cell proliferation, apoptosis, and bone destruction. These three biological processes are related to the structure of cholesteatoma. Cholesteatoma is histologically divided into three parts: the cystic content, matrix, and perimatrix. The cystic content is the exfoliated debris of epithelial cells after keratinization, the matrix is composed of epithelial cells, and the perimatrix is composed primarily of fibroblasts and capillaries. Abnormal proliferation and apoptosis are characteristics of epithelial cells of the matrix, whereas the secreted cytokines of the perimatrix are involved in bone destruction [3,4]. Recent studies have revealed that these specific cholesteatoma phenotypes, compared to those of the normal epithelium, are primarily caused by transcriptional regulation; thus, recent studies on the pathogenesis of cholesteatoma have focused on transcriptional regulation.
Studies have shown that mammalian transcription is mainly regulated by non-coding RNAs (ncRNAs), which have emerged as a popular research topic after the completion of the Human Genome Project. Although ncRNAs are not involved in protein translation, they are abundant in the genome, with only approximately 2% of genes encoding proteins and the remaining approximately 98% encoding ncRNAs [5]. ncRNAs are classified by length, into small ncRNAs (which include but are not limited to microRNA [miRNA]) and long ncRNA (lncRNA), and they are further classified into regulatory ncRNAs and non-regulatory ncRNAs, mainly by function. Regulatory ncRNAs include miRNA, small interfering RNA (siRNA), PIWI-interacting RNA (piRNA), lncRNA, and circular RNA (circRNA). Non-regulatory ncRNAs, also known as housekeeping ncRNAs, include ribosomal RNA (rRNA), tRNA-derived small RNA (tsRNA), small nuclear RNA (snRNA), and small nucleolar RNA (snoRNA) and mainly function as genetic structural elements to ensure genomic stability [6,7]. Because of their key roles in gene expression, cell cycle regulation, and the development of many cancers, regulatory ncRNAs, especially miRNAs, lncRNAs, and circRNAs, have received increasing attention [8].
miRNAs are a class of small, evolutionarily highly conserved, single-stranded ncRNAs, ranging from 18 to 25 nucleotides in length [9,10]. They are processed from primary transcription products in the nucleus by the Drosha enzyme into hairpin RNAs, which are transported from the nucleus to the cytoplasm with the help of the exportin-5 complex, and finally further cleaved by Dicer to form functional miRNAs [11,12]. Functional miRNAs bind the 3′ untranslated region (UTR) of the target gene with complete or incomplete complementarity, thus degrading the target mRNA or inhibiting its translation to regulate gene expression.
lncRNAs are ncRNAs greater than 200 nucleotides in length that contain miRNA responsive elements (MREs). lncRNAs have been studied extensively in the regulation of gene expression at the epigenetic, transcriptional, and post-transcriptional levels [13]. A major function of lncRNAs is to serve as an “miRNA sponge” through the binding of its MREs to miRNAs, thus competitively inhibiting the transcriptional regulation of miRNAs [14].
CircRNAs are ncRNAs with a closed-loop structure [15] formed by an interconnection of the 5′ and 3′ ends of linear RNAs, which makes them immune to the activity of RNA exonucleases; thus, they remain stable during gene expression [16]. Unlike linear RNAs, circRNAs form covalently closed continuous loops and act as gene regulators in mammals. They act as miRNA “sponges” to adsorb miRNAs and thereby inhibit their activity [17], enter exosomes and participate in cellular communication [18], and are even involved in RNA–protein interactions in some cases [19]. However, their main function is associated with their abundant miRNA-binding sites, making them important components of the competing endogenous RNA (ceRNA) network.
In 2011, Salmena et al. [20] proposed the ceRNA hypothesis. Specifically, ceRNA does not refer to a specific type of RNA, but rather a post-transcriptional regulatory mechanism that involves the inhibition of miRNA-induced silencing complex formation through binding to MREs, which results in increased mRNA expression downstream of the miRNA. Recent studies have found that both protein-coding mRNAs and non-coding lncRNAs, circRNAs, and pseudogenes have ceRNA-based regulatory mechanisms. The core of this regulatory mechanism is the MRE, and various types of RNAs that contain MREs bind to miRNAs because multiple different RNAs can contain the same MREs. Thus, these RNAs compete with each other to bind miRNAs and are also known as miRNA sponges [21]. The 3′ UTR of protein-encoding mRNAs contains MREs that can bind to miRNAs in a fully or imperfectly complementary manner [22], which can result in the degradation of mRNAs or can affect their translation, reducing the expression of their encoded target proteins [23–25]. ncRNAs containing the same or different MREs for the same miRNA, such as lncRNA, circRNA, or pseudogenes, can compete for miRNAs like a sponge through the ceRNA mechanism, such that the amount of miRNAs that bind mRNAs is significantly reduced, thus relieving or attenuating the inhibitory effect of miRNAs on mRNA translation. Thus, higher ceRNA content leads to increased expression of the corresponding mRNA-encoded protein, and vice versa. Of course, mRNA itself can also regulate other mRNA molecules (circRNA, lncRNA) in a trans manner via these binding regions, exerting a ceRNA regulatory effect [26]. For example, Tay et al. [27] found that vesicle-associated membrane protein-associated protein A (VAPA) and CCR4-NOT transcription complex subunit 6-like (CNOT6L) mRNAs have the same MREs as PTEN and can act as ceRNA to competitively bind miRNAs, thereby indirectly altering PTEN (phosphatase and tensin homolog deleted on chromosome 10) abundance and suppressing tumorigenesis. This article reviews the research on the roles of these three types of ncRNAs in cholesteatoma, in accordance with the ceRNA hypothesis (Table 1) [28–42].
All currently known ncRNAs in cholesteatoma of the middle ear
miRNA AND CHOLESTEATOMA
Overview of miRNA
miRNA was first reported in 1993 by Lee et al. [43], who studied lin-4, a Caenorhabditis elegans gene that regulates nematode development; they found that the gene does not encode a protein, but instead forms two short RNA molecules less than 70 nucleotides in length. One of these precursor RNAs, approximately 20 nucleotides in length, is the antisense molecule that is complementary to the 3′ UTR of the lin-14 gene, and it was determined that it significantly reduces the protein expression of lin-14. Since then, more researchers have studied miRNAs in different species and diseases, and it was eventually found that miRNAs are widely present in various species and play important regulatory roles in a variety of biological mechanisms.
Currently, miRNAs are considered a class of single-stranded RNAs of approximately 22 nucleotides in length, formed via the transcription of genomic DNA in the nucleus, cleavage, and then transport to the cytoplasm for further processing. Initially, the DNA encoding the miRNA is transcribed by RNA polymerase II to form a hairpin structure, the primary miRNA (pri-miRNA) [44], which is characterized by a hairpin structure with a 7MGpppG cap and a double-stranded polyadenylate tail (AAAA). pri-miRNA is recognized and bound by the DGCR8 protein in the nucleus and is subsequently cleaved by the Drosha enzyme to form a precursor miRNA (pre-miRNA) containing a stem-loop structure approximately 70 nucleotides in length [45]. pre-miRNA is then recognized by exportin 5 and transported to the cytoplasm [46]. In the cytoplasm, pre-miRNA is cleaved by the Dicer enzyme into a double-stranded miRNA molecule of approximately 22 nucleotides, which in turn is opened via the activity of the AGO2 protein. One of the strands becomes a mature miRNA, also known as the guide strand, which enters the RNA-induced silencing complex [47] and binds to the target mRNA to silence its expression. Nucleotides 2–8 of the 5′ end of the miRNA are known as the seed sequence, which can complementarily bind the 3′ UTR of the target mRNA. Depending on its degree of complementary pairing, miRNA affects mRNA to different degrees. Complete complementarity can result in degradation of the target mRNA, whereas incomplete pairing can inhibit mRNA translation, thus affecting the protein expression level [48,49]. Recent studies have shown that miRNAs can also complementarily pair with the 5′ terminal non-coding region or open reading frames of target mRNAs [50–52].
Involvement of miRNA in the development and progression of cholesteatoma and its mechanism of action
miR-34a and cholesteatoma of the middle ear
In cholesteatoma cells and a BALB/C nude mouse model, Zheng et al. [28] found lower miR34a expression in cholesteatoma cells than in external ear canal skin tissue using quantitative real-time polymerase chain reaction (qRT-PCR). They also successfully prepared and delivered the mir34a small molecule regulator, rubine, by nanotechnology, into the cells. Rubine can passively target cells and regulate miR-34a. The researchers observed an altered cell phenotype in cholesteatoma cells and model mice, specifically the pro-apoptotic inhibition of cell proliferation and migration; they also found that miR34 induced changes in the expression levels of Bcl-2, Cdk6, and cyclin D1, which were dependent on the PTEN/P13K/protein kinase B (Akt) pathway (Fig. 1A).

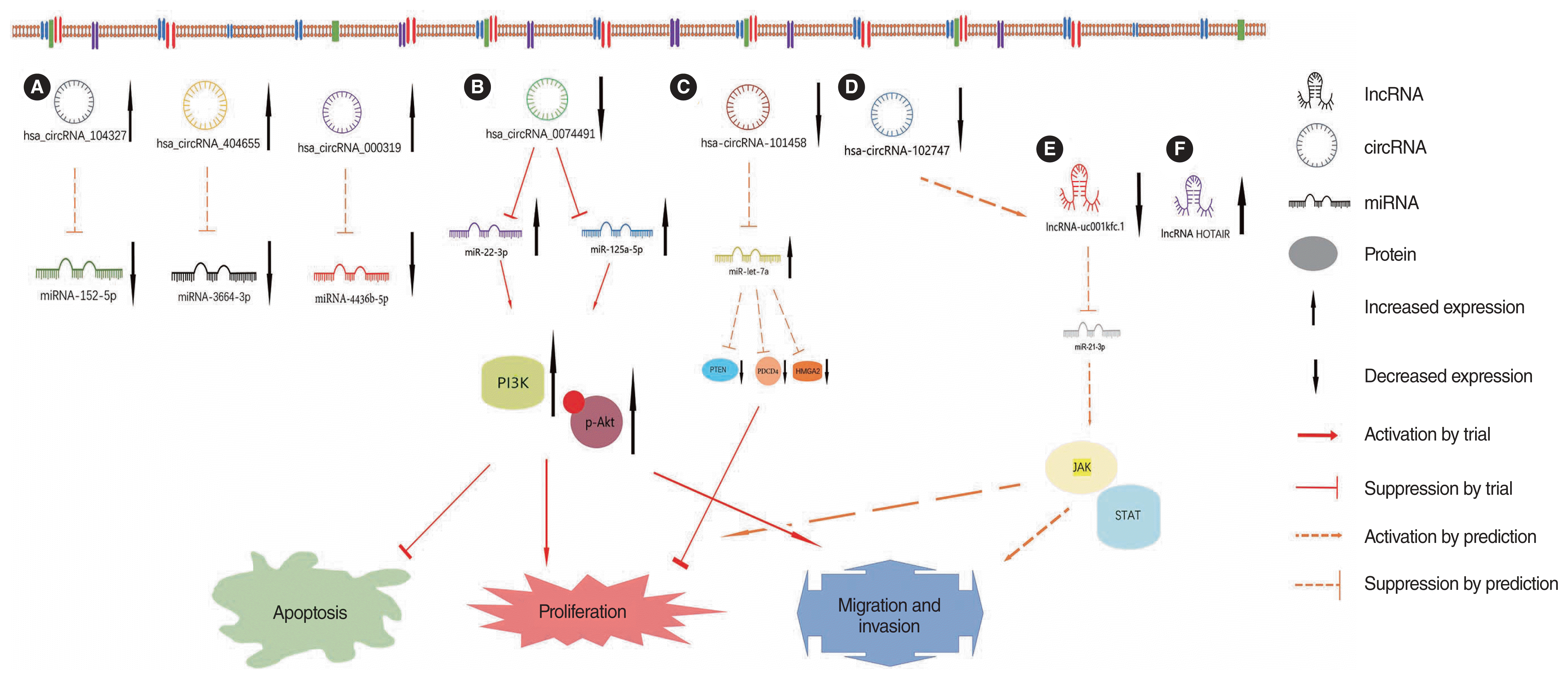
Summary of the main miRNA and molecular mechanisms involved in middle ear cholesteatoma. (A) miR-34a inhibits the proliferation and migration of cholesteatoma cells, promotes the apoptosis of cholesteatoma cells by targeting Bcl-2, Cdk6, cyclin D1, and negatively regulates the PTEN/PI3K/AKT signaling pathway. (B) miR-21, which is a downstream target of CD14, IL-6R, gp130, and STAT3, promotes proliferation and invasion and inhibits apoptosis in cholesteatoma cells by negatively regulating PTEN, PDCD4 and HMGA2. (C) miR-let-7a inhibits cholesteatoma cell proliferation and invasion and promotes their apoptosis, and this biological function might be achieved by negatively regulating miR-21 expression. (D) miR-203a affects p-Akt levels by targeting Bmi1; promoting cholesteatoma proliferation, colony formation, and migration; and inhibiting apoptosis. (E) miR-21-3p and miR-16-1-3p expression are significantly elevated in middle ear cholesteatoma tissues, whereas miR-10a-5p expression is significantly decreased. It is hypothesized that miR-16-1-3p and miRNA-10a-5p might induce cholesteatoma tissue hyperproliferation and regulate cholesteatoma formation through the PI3K-Akt signaling pathway. (F) NF-κB activation by TNF-α, IL-1β, and IL-6 during the development and progression of middle ear cholesteatoma increases miR-802 expression, which in turn promotes cell proliferation, and this is accomplished by downregulating the PTEN/p-AKT pathway. (G) miR-142-5p has a direct negative regulatory effect on CDK5, which is involved in regulating the secretion of inflammatory cytokines, such as TNF-α, TGF-β1, IL-5, IL-6, and IL-17A, in cholesteatomas. (H) Downregulation of exosomal miR-106b-5p derived from cholesteatoma perimatrix fibroblasts promotes angiogenesis via Angpt2 overexpression. miR, microRNA; PTEN, phosphatase and tensin homolog deleted on chromosome 10; PI3K, phosphoinositide 3-kinase; p-Akt, phosphorylated protein kinase B; Bcl-2, B-cell lymphoma-2; Cdk6, cyclin-dependent kinases 6; IL-6R, interleukin 6 receptor; CD14, lipopolysaccharide 14; STAT3, signal transducer and activator of transcription 3; gp130, glycoprotein 130; PDCD4, programmed cell death protein 4; HMGA2, high mobility group AT-hook 2; Bmi1, B cell-specific Moloney murine leukemia virus insertion site 1; TNF, tumor necrosis factor; IL, interleukin; NF-κB, nuclear factor kappa B; CDK5, cyclin-dependent kinase 5; TGF, transforming growth factor; miRNA, microRNA; Angpt2, angiopoietin 2.
miR-21 and cholesteatoma of the middle ear
Friedland et al. [29] extracted RNA and protein from cholesteatoma tissue and normal skin samples of six patients taken at the time of surgery. They used qRT-PCR to assess the levels of human miRNA, and western blot analysis was used to assess the levels of downstream target proteins. The results revealed that miRNA-21 (hsa-miR-21) was expressed at a 4.4-fold higher level in cholesteatoma tissues than in normal skin (P=0.0011). The expression levels of PTEN and PDCD4 (programmed cell death protein 4), which are downstream targets of hsa-miR-21, were found to be significantly reduced in three of the four cholesteatoma samples and negatively correlated with hsa-miR-21 expression. In contrast, the upstream regulators (CD14, interleukin 6 receptor [IL-6R], gp130, and signal transducer and activator of transcription 3 [STAT3]) believed to contribute to hsa-miR-21 expression were all determined to be present in cholesteatoma tissues. PTEN and PDCD4 act as oncogenes, and these translated proteins were shown to control aspects of apoptosis, proliferation, invasion, and migration. It was hypothesized that the aberrantly elevated expression of hsa-miR-21 in cholesteatoma decreases PTEN and PDCD4 expression, which promotes proliferation and invasion and inhibits apoptosis in cholesteatoma cells (Fig. 1B).
Chen et al. [30] performed cell culture of 30 surgical cholesteatoma specimens, transfected cells with a miR-21 mimic, miR-21 inhibitor, or empty plasmid, and plotted the resulting growth curves. The miR-21 expression levels in the three groups were determined by qRT-PCR, the proliferation and apoptosis of cholesteatoma keratinocytes were determined by EdU and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining, respectively, and their invasive ability was evaluated using 6-well Transwell assays. Compared to the miR-21 inhibitor or empty plasmid group, miR-21 expression was significantly upregulated in cholesteatoma keratinocytes transfected with the miR-21 mimic. The number of EdU-positive cells, reflecting cell proliferation, also significantly increased, whereas the number of TUNEL-positive cells, reflecting apoptosis, significantly decreased. In addition, migrating cells, based on the Transwell assay, were more abundant in the miR-21 mimic group than in the miR-21 inhibitor or empty plasmid group. The researchers concluded that miR-21 promotes keratinocyte proliferation and invasion in cholesteatomas and inhibits their apoptotic capacity.
miR-106b and cholesteatoma of the middle ear
Li et al. [31] extracted miR-106b-5p exosomes from the cholesteatoma perimatrix and found that they could promote the proliferation, migration, and angiogenesis of human umbilical vein endothelial cells, concluding that the downregulation of miR-106b-5p expression can promote angiogenesis in tissues surrounding cholesteatomas. Dual-luciferase assays showed that this process was induced by miR-106b-5p activating angiopoietin 2. Finally, it was concluded that exosomes derived from human cholesteatoma perimatrix fibroblasts transport miR-106b-5p, expressed at low levels, to endothelial cells, promoting angiogenesis via angiopoietin 2 overexpression (Fig. 1H).
miR-203a and cholesteatoma of the middle ear
Zang et al. [32] used qRT-PCR and western blotting to detect the miRNA, mRNA, and protein levels of miR-203a, Bmi1, and phosphorylated Akt (p-Akt) and immunohistochemical staining to observe the expression and distribution of Bmi1 and p-Akt in the skin behind the ear of control subjects and in cholesteatoma samples. Dual-luciferase reporter gene assays were used to analyze the relationship between miR-203a and Bmi1. Both miR-203a and Bmi1 were transfected into immortalized human keratinized cell lines (HaCaT cells), and their role in cell proliferation, apoptosis, and migration was investigated. The results showed that miR-203a expression was downregulated in cholesteatoma tissues, whereas Bmi1 expression was upregulated. Dual-luciferase reporter gene assays showed that Bmi1 was a direct target of miR-203a. miR-203a silencing increased Bmi1 expression, promoted the proliferation, colony formation, and migration of HaCaT cells, and inhibited apoptosis. The expression of p-Akt was significantly elevated in cholesteatoma tissues and positively correlated with Bmi1. The expression of Bmi1 protein and the p-Akt protein in the Bmi1 siRNA group were significantly lower than in the control siRNA group. When cells were co-transfected with Bmi1 siRNA and a miR-203a inhibitor, the expression of the Bmi1 and p-Akt proteins significantly recovered. Ultimately, the researchers concluded that miR-203a affects p-Akt levels by regulating Bmi1; promoting cholesteatoma proliferation, colony formation, and migration; and inhibiting apoptosis (Fig. 1D).
miRNA-16-1-3p and miRNA-10a-5p and cholesteatoma of the middle ear
Xie et al. [33] used microarrays to sequence five pairs of specimens and found 44 upregulated miRNAs (including miRNA-21-3p, miRNA-584-5p, and miRNA-16-1-3p) and 175 downregulated miRNAs (including miRNA-10a-5p, miRNA-152-5p, and miRNA-203b-5p) in cholesteatoma tissues (fold change [FC] ≥2.0, P<0.05). qRT-PCR confirmed that miRNA-21-3p and miRNA-16-1-3p expression was significantly elevated in middle ear cholesteatoma tissues, whereas miRNA-10a-5p expression was significantly reduced. Gene Ontology (GO) classification (http://geneontology.org/) and Kyoto Encyclopedia Genes and Genomes (KEGG) pathway analysis (https://www.genome.jp/kegg/expression) suggested that the target genes play roles in protein serine/threonine phosphatase inhibitor activity, glucocorticoid receptor binding, and activin binding in terms of their molecular function. For biological processes, the target genes were involved in 831 biological processes, including the regulation of viral protein levels in host cells, negative regulation of bone resorption, and histone mRNA catabolic processes. Regarding cellular components, most target genes were part of several complexes, such as the Set1C/COMPASS, PRC1, and ESC/E (Z) complexes. In addition, KEGG pathway analysis revealed that the putative target genes were involved in 39 signaling pathways, including the p53, PI3K-Akt, and osteoclast differentiation signaling pathways. Finally, it was hypothesized that miRNA-16-1-3p and miRNA-10a-5p might induce cholesteatoma tissue hyperproliferation and regulate cholesteatoma formation through the PI3K-Akt signaling pathway (Fig. 1E).
miR-let-7a and cholesteatoma of the middle ear
Zhang et al. [34] collected 20 postoperative cholesteatoma specimens for cell culture and transfected them with a miR-let-7a mimic, miR-let-7a inhibitor, or empty plasmid. qRT-PCR was used to detect miR-let-7a and miR-21 expression levels in the three groups. The cell cycle status, proliferation, and apoptosis of cholesteatoma cells were determined by flow cytometry, EdU, and TUNEL staining, respectively, and their invasive ability was determined by 6-well Transwell assays. The results showed that miR-let-7a expression was significantly upregulated in the cholesteatoma cells transfected with the miR-let-7a mimic compared to that in the miR-let-7a inhibitor or empty plasmid group, and flow cytometry showed that more cells in the miR-let-7a mimic group were in the G0/G1 phases than in the G2/M or S phases, consistent with the cell cycle status of the two groups. The number of EdU-positive cells, reflecting proliferation, was also significantly reduced, whereas TUNEL-positive cells, reflecting apoptosis, were significantly increased. Transwell assays showed that the number of migrating cells in the miR-let-7a inhibitor or empty plasmid group was also higher than that in the miR-let-7a mimic-transfected group (Fig. 1C). In addition, miR-let-7a and miR-21 expression showed significant negative correlations in the three groups. It was concluded that miR-let-7a could inhibit cholesteatoma cell proliferation and invasion and promote their apoptosis, and the researchers proposed that this biological function might be achieved by regulating miR-21 expression.
Chen et al. [35] collected postoperative cholesteatoma specimens from 14 adult patients and 13 pediatric patients and corresponding normal skin tissue from each patient. qRT-PCR was used to detect miR-let-7a and miR-21 expression, and western blotting was used to detect protein levels of PTEN, PDCD4, and HMGA2 (high mobility group AT-hook 2) in each sample. The expression of miR-let-7a and miR-21 in cholesteatoma tissues was significantly higher than that in normal skin tissues, and their expression was more pronounced in children than in adults; the difference was statistically significant. There were no significant differences in expression between children and adults in normal skin tissues. PTEN, PDCD4, and HMGA2 expression was significantly lower in cholesteatoma tissues than in normal skin tissues, especially in children, but there was no difference in the expression of these proteins between children and adults in normal skin tissues. It was concluded that increased miR-let-7a and miR-21 expression might be associated with a more destructive clinical presentation of cholesteatoma in children than in adult patients, and this biological behavior might be achieved by regulating the downstream expression of PTEN, PDCD4, and HMGA2 proteins.
miR-802 and cholesteatoma of the middle ear
Li and Qin [36] collected intraoperative cholesteatoma specimens and corresponding posterior ear skin tissues from patients, treated keratinocytes with tumor necrosis factor (TNF)-α (10 ng/mL), interleukin (IL)-1β (20 ng/mL), and IL-6 (20 ng/mL) for 6 hours, and used the keratinocytes to establish a miR-802 mimic, miR-802 inhibitor, and empty plasmid group. TNF-α, IL-1β, IL-6, PTEN, p-Akt, and miR-802 expression in cholesteatoma tissue, normal epithelial tissue, and treated cells was detected by western blotting and qRT-PCR. Chromatin immunoprecipitation (ChIP) was used to analyze intracellular p65 and miR-802 co-precipitation. Flow cytometry and BrdU staining were used to detect cell proliferation and the cell cycle status, respectively. The miRWalk, TargetScan, RNAhybrid, and miRanda software programs were used to predict mRNAs downstream of miR-802, and the prediction results were verified by dual-luciferase assays.
The results revealed that TNF-α, IL-1β, and IL-6 expression was significantly higher in cholesteatoma than in normal epithelial tissue. miR-802 expression was also significantly higher in keratinocytes treated with TNF-α, IL-1β, and IL-6 than in normal skin. The ChIP results showed that p65 was associated with the miR-802 promoter. Flow cytometry results showed that compared with that in normal control transfected cells, the proportion of G1/G0-phase cells was significantly lower, and the proportion of S-phase cells was higher in the miR-802 mimic group than in the miR-802 inhibitor group, with opposite results observed for the miR-802 inhibitor group. BrdU staining showed significantly more positive cells in the miR-802 mimic group than in the other two groups. Multiple software programs predicted PTEN as the downstream mRNA of miR-802, and dual-luciferase assays verified this targeting relationship. The expression of PTEN in the miR-802 mimic group was lower than that in the other two groups, whereas the expression of p-AKT was higher than that in the other two groups. It was concluded that the miR-802 promoter contains a functional nuclear factor kappa B (NF-κB)/P65-binding site and that high miR-802 expression promotes keratinocyte proliferation and differentiation. Furthermore, PTEN was found to be a direct downstream target mRNA of miR-802. In conclusion, NF-κB activation during the development and progression of middle ear cholesteatoma increases miR-802 expression, which in turn promotes cell proliferation, and this is accomplished by downregulating the PTEN/p-AKT pathway (Fig. 1F).
miR-142-5p and cholesteatoma of the middle ear
Sui et al. [37] collected 20 cholesteatoma specimens and 20 normal skin tissue samples, cultured human keratinocytes, stimulated them with lipopolysaccharide (LPS), and transfected them with CDK5 siRNA, an miR-142-5p mimic, or an miR-142-5p inhibitor. Next, immunohistochemistry, immunofluorescence, western blotting, and qRT-PCR were used to detect the expression of miR-142-5p and CDK5 in the pairs of tissues and the treated and transfected cells. Dual-luciferase assays were used to confirm the direct targeting relationship between miR-142-5p and CDK5. Finally, enzyme-linked immunosorbent assays and qRT-PCR were used to detect TNF-α, transforming growth factor (TGF)-β1, IL-5, IL-6, and IL-17A expression levels in tissue specimens and treated cells.
CDK5 expression was significantly higher in acquired cholesteatoma and LPS-stimulated human HaCaT keratinocytes than in normal skin and stimulated cells, whereas miR-142-5p expression was significantly lower than in normal tissues and cells; the negative correlation between the two was statistically significant. In addition, the dual luciferase reporter gene assay confirmed that miR-142-5p directly targets the 3′-UTR of CDK5. qRT-PCR revealed elevated levels of TNF, TGFB1, IL5, IL6, and IL17A in acquired cholesteatoma, which were positively correlated with CDK5 expression. In transfected cells, CDK5 knockdown significantly reduced LPS-induced inflammatory cytokine secretion, and the miR-142-5p inhibitor significantly increased the effects of CDK5 knockdown on inflammatory cytokine expression and secretion. It was concluded that miR-142-5p has a direct negative regulatory effect on CDK5, CDK5 is involved in regulating inflammatory cytokine secretion in cholesteatoma, and miR-142-5p is involved in the regulation of inflammation in cholesteatoma via the CDK5-mediated inflammatory pathway (Fig. 1G).
lncRNAs AND CHOLESTEATOMA
Overview of lncRNAs
lncRNAs are ncRNAs greater than 200 nucleotides in length that can be transcribed by RNA polymerase II and depend on post-transcriptional modifications such as 5′ end-capping and 3′ end-polyadenylation. The origin of lncRNAs is unclear, but there are four prevailing views [5,53,54] as follows: (1) mutation, according to which a protein-coding gene sustains a mutation that produces a frameshift mutation in the open reading frame when the DNA is transcribed into a coding RNA, thus forming a new RNA; (2) recombination, in which multiple non-transcribed and separate gene sequences are recombined, allowing several non-transcribed and originally distant sequence regions to be merged, producing a multi-exon lncRNA; (3) duplication, according to which duplication of adjacent structural units in the ncRNA sequence, creating a longer lncRNA transcript; (4) insertion, in which a transposable element containing a transcription start site is inserted into the genome to produce a functional lncRNA that is combined with the previously coded sequence. The classification of lncRNAs is not uniform and has multiple premises. Based on the relative positions of lncRNA coding sequences and a protein-coding gene, they can be classified into the following five categories [55–59]: (1) sense lncRNA: lncRNA sequences in the same direction as the sense strand of the protein-coding gene; (2) antisense lncRNA: lncRNA sequences in the same direction as the antisense strand of the protein-coding gene; (3) bidirectional lncRNA: lncRNA sequences located on the opposite strand from the protein-coding gene; (4) intronic lncRNA: lncRNA sequences completely derived from the introns of another transcript; and (5) intergenic lncRNA: lncRNA sequences not located near any other protein-coding gene loci. lncRNAs play multiple regulatory roles [60,61]—namely, regulation at the transcriptional, epigenetic, and post-transcriptional levels based on interactions with DNA, RNA, or proteins.
Regulation at the transcriptional level
lncRNAs can silence genes by directly interfering with transcription factor binding to the promoters of protein-coding genes, thereby preventing their expression [62]. For example, the transcription of lncRNAs upstream of SER3 interferes with the binding of RNA polymerase II to DNA, thus inhibiting SER3 expression [63].
Further, lncRNAs inactivate transcription factors. McHugh et al. [64] showed that the lncRNA XIST silences transcription factors by directly interacting with SHARP, recruiting SMRT, activating HDAC3, and deacetylating histones, thereby excluding Pol II from the X chromosome. Recent studies have indicated that lncRNAs have significant effects on gene expression based on neighboring promoter activity, including transcription initiation and splicing processes. Engreitz et al. [65] analyzed 12 genomic loci producing lncRNAs using genetic manipulation and found that five loci affected the expression of neighboring genes in a cis manner. None of these effects required specific lncRNA transcripts per se, but rather involved general processes associated with their production, including the enhancer-like activity of gene promoters, transcriptional processes, and splicing of transcripts. These results suggest that crosstalk between neighboring genes is a general phenomenon involving multiple mechanisms and cis-regulatory signals, including a novel role for RNA splice sites. These mechanisms could explain the function and evolution of some genomic loci that produce lncRNAs and are broadly involved in the regulation of coding and non-coding genes.
Regulation at the epigenetic modification level
Epigenetic modifications include histone [66] and DNA methylation [67], histone acetylation, and ubiquitination [68]. Many lncRNAs can regulate chromatin [69], but the mechanisms by which they target genomic targets are unknown. Yu et al. [70] found that the lncRNA XIST is consistently required to silence a subset of X-linked immune genes, such as TLR7, in adult human B cells. XIST-dependent genes lack promoter DNA methylation and require sustained XIST-dependent histone deacetylation. XIST RNA-directed proteomics and CRISPRi screens revealed unique somatic cell type-specific XIST complexes and identified TRIM28, which mediates Pol II pausing at X-linked gene promoters in B cells. These results suggest that lncRNA–protein complexes may play a substantial role in sex-based differences in biology and medicine through epistatic modifications. Wan et al. [71] found that lncRNA-JADE was induced after DNA damage in an ataxia-telangiectasia mutated-dependent manner. lncRNA-JADE transcriptionally activates Jade1, a key component of the human acetylase binding to ORC1 (HBO1) histone acetylation complex. Consequently, lncRNA-JADE induces histone H4 acetylation in the DNA damage response. lncRNA-JADE levels were observed to be markedly higher in human breast tumors than in normal breast tissue samples. lncRNA-JADE knockdown significantly inhibited breast tumor growth in vivo. These results suggest that lncRNA-JADE is a key functional link between the DNA damage response and histone H4 acetylation and that lncRNA-JADE dysregulation might contribute to breast tumorigenesis.
Regulation at the post-transcriptional level
lncRNAs regulate various aspects of post-transcriptional mRNA function, similar to small ncRNAs, such as miRNA and snoRNA, which usually involves complementary base pairing with target mRNAs [72,73]. Cui et al. [74] quantitatively demonstrated, by immunohistochemical staining and qRT-PCR, that complementary base pairing between the highly upregulated in liver cancer (HULC) lncRNA and the 5′-UTR of CLOCK mRNA underlies the regulation of CLOCK expression by the HULC lncRNA, which interferes with circadian rhythms by upregulating the circadian oscillator clock in hepatocellular carcinoma cells, thereby promoting hepatocarcinogenesis. It was concluded that lncRNA accelerates hepatocarcinogenesis by interfering with its circadian rhythm.
Involvement of lncRNAs in the development and progression of cholesteatoma and associated mechanism
The HOTAIR lncRNA and cholesteatoma of the middle ear
Li et al. [38] analyzed six lncRNAs that are closely associated with cholesteatoma epithelial keratinocyte differentiation and apoptosis according to an extensive literature review. The expression of these six lncRNAs (HOTAIR, ANCR, TINCR, PRINS, BANCR, PICSAR) in 25 middle ear cholesteatoma epithelial and 15 normal outer ear canal skin tissue samples was compared. The results showed that HOTAIR expression levels in cholesteatoma epithelial tissues were significantly upregulated (Fig. 2) compared to those in outer ear canal skin tissue (P<0.01), whereas the differences in ANCR, TINCR, PRINS, BANCR, and PICSAR expression between the two tissues were not statistically significant (P>0.05). One-way analysis of variance was performed for the expression levels of HOTAIR in patients with stage I, II, and III cholesteatoma, and no statistically significant differences among patients with different degrees of cholesteatoma were found (P>0.05).
Summary of the main circRNA/lncRNA and molecular mechanisms involved in middle ear cholesteatoma. (A) hsa_circRNA_104327 and hsa_circRNA_404655 expression is significantly higher and hsa_circRNA_000319 expression is significantly lower in middle ear cholesteatoma than in normal skin tissue. These circRNAs have been found to interact with miRNA-152-5p, miRNA-3664-3p, and miRNA-4436b-5p, respectively, according to the ceRNA hypothesis. (B) hsa_circ_0074491 plays a key role in facilitating cell proliferation, migration, and invasion and repressing cell apoptosis in cholesteatoma through inactivating the PI3K/Akt pathway via competitively binding to miR-22-3p and miR-125a-5p. (C) hsa-circRNA-101458 expression has been confirmed to be significantly lower in cholesteatoma than in epithelial tissues. It could inhibit proliferation by competitively interacting with miR let-7a-3p according to ceRNA network prediction analysis. (D) hsa-circRNA-102747 has been verified to be significantly lower in cholesteatoma by qRT-PCR. According to one hypothesis, circRNA-102747/lncRNA-uc001kfc.1/miR-21-3p/targeted mRNAs might regulate malignant characteristics according to ceRNA network prediction analysis. (E) lncRNA-uc001kfc.1 expression has been confirmed to be significantly lower in cholesteatoma tissues. It is predicted that lncRNA-uc001kfc.1 could regulate the expression of major nodal proteins of the JAK/STAT pathway through miR-21 to alter the proliferative and invasive behavior of cholesteatoma cells. (F) Expression levels of the HOTAIR lncRNA in cholesteatoma tissues are significantly upregulated compared to that in outer ear canal skin tissue. circRNA, circular RNA; miRNA, microRNA; PI3K, phosphoinositide 3-kinase; p-Akt, phosphorylated protein kinase B; PTEN, phosphatase and tensin homolog deleted on chromosome 10; PDCD4, programmed cell death protein 4; HMGA2, high mobility group AT-hook 2; lncRNA, long ncRNA; JAK, Janus kinase; STAT, signal transducer and activator of transcription; ceRNA, competing endogenous RNA; qRT-PCR, quantitative real-time polymerase chain reaction.
lncRNA-uc001kfc.1 and cholesteatoma of the middle ear
Gao et al. [39] conducted a microarray analysis to profile differences in lncRNA and mRNA expression between four pairs of cholesteatoma and matched normal skin samples. According to this profiling data, 11,815 lncRNAs and 7,692 mRNAs were detected. The lncRNAs were classified into six categories: bidirectional (4.46%), exon sense-overlapping (1.16%), intron sense-overlapping (3.47%), natural antisense (8.75%), intronic antisense (12.71%), and intergenic (69.47%). With threshold FC ≥2.0 and a P<0.05, the researchers identified 787 lncRNAs and 591 mRNAs that were differentially expressed between cholesteatoma and matched normal skin tissues. In cholesteatoma samples, the levels of 181 lncRNAs and 155 mRNAs were upregulated (FC ≥2.0, P<0.05) and those of 606 lncRNAs and 436 mRNAs were downregulated (FC≥2.0, P<0.05) compared to those in normal skin samples. The microarray profile and RNA sequencing datasets were deposited into the Gene Expression Omnibus (GEO) with accession number GSE102673.
Next, enrichment analysis was performed. The upregulated and downregulated mRNAs were analyzed separately, and the top 10 enriched GO terms were listed, including biological processes, cellular components, and molecular functions. The most enriched biological processes associated with the upregulated mRNAs included the regulation of phosphatidylinositol 3-kinase signaling (GO:0014066), phosphatidylinositol 3-kinase signaling (GO:0014065), and positive regulation of phosphatidylinositol 3-kinase signaling (GO:0014068). Phosphatidylinositol 3-kinase binding (GO:0043548) was the most significantly enriched function based on the upregulated mRNAs in the molecular function analysis. The KEGG pathway enrichment analysis (http://www.genome.jp/kegg/pathway.html) for differentially expressed mRNAs showed that the most enriched pathways were involved in bacterial invasion of epithelial cells (hsa05100), viral myocarditis (hsa05416), and hepatitis B (hsa05161).
Among these significantly differentially expressed lncRNAs (ENST00000415386; ENST00000420253; NR_024468; T044224; T347175; and uc001kfc.1) were selected for qRT-PCR validation. lncRNA-uc001kfc.1 expression was confirmed to be significantly abnormal in cholesteatoma tissues (Fig. 2E). The miRBase prediction of the relationship between lncRNAs and mRNAs, the miRcode prediction of the relationship between lncRNAs and miRNAs, and miRanda (www.microRNA.org/) and TargetScan predictions of the relationship between miRNAs and mRNAs showed that lncRNA-uc001kfc.1 regulates the expression of major nodal proteins of the JAK/STAT pathway through miR-21, thereby altering the proliferative and invasive behavior of cholesteatoma cells.
circRNAs AND CHOLESTEATOMA
Overview of circRNAs
The first discovery of human circRNA was in 1976 when Sanger et al. [75] identified a pathogenic, single-stranded circular viroid in a study of potato spindle tuber disease. Three years later, Hsu and Coca-Prados [76] observed circRNAs in the cytoplasm of eukaryotic cells by electron microscopy. In 1993, Cocquerelle et al. [77] identified circRNAs in human somatic cells. With the rapid development of high-throughput sequencing, in 2013, Memczak et al. [78] sequenced and computationally analyzed human, mouse, and nematode RNAs to systematically investigate circRNAs and detected thousands of well-expressed, stable circRNAs, which often showed tissue/developmental stage-specific expression. Sequence analysis indicated that circRNAs carry out important regulatory functions. Systematic studies on circRNAs have since been conducted worldwide.
Formation of circRNAs
circRNAs are classified into three categories based on their origin: exonic circRNAs (ecircRNAs), circular intron circRNAs (ciRNAs), and exon-intron circRNAs (eiciRNAs) [79]. Using a novel identification method, Liu et al. [80] found that some circRNAs can originate from exons, introns, and intergenic regions in humans, mice, and rice and are known as interior circRNAs. These circRNAs are formed from the mRNA precursor pre-mRNA via nonlinear reverse splicing, a process that depends on cis-acting elements and trans-acting factors [81].
circRNAs differ from linear ncRNAs
First, circRNAs are widely found in different tissues of eukaryotic organisms, such as the stomach [82,83], pancreas [84], liver [85,86], human and mouse brain [87–89], mammary gland [90,91], prostate [92], and thyroid [93,94]. A distinctive feature of circRNAs is their highly conserved sequences. Conservation describes how a range of macromolecules are perfectly preserved throughout biological evolution. circRNAs are highly evolutionarily conserved across species [87,95–97]. Rybak-Wolf et al. [88] used the LiftOver tool to determine whether neuronal circRNA expression is conserved between mammals using genome-wide comparisons and found that 4,522 of 15,849 mouse circRNAs are conserved in humans. This conservation increases with increasing expression. For an additional 4,527 mouse circRNAs, they observed overlapping human circRNAs with one identical splice site and one different splice site, suggesting that splice sites compete in circRNA synthesis, which could be explained by the gain or loss of complementary elements in the surrounding introns. For 5,278 mouse circRNAs, no human homolog was detected, whereas for 1,522 circRNAs, they were unable to map splice sites to the human genome. In addition, they found through Sanger sequencing that head-to-tail linkage sequences are precisely conserved in humans and mice.
Regarding tissue specificity, there are significant differences in circRNA expression. For example, sequencing of the digestive and respiratory systems revealed significant differences in circRNA expression [98,99]. Therefore, tissue specificity could make circRNAs diagnostic factors for tissue-specific diseases [100].
In terms of stability, circRNAs are circular structures without 5′ and 3′ ends, and thus, they are insensitive to RNAses and more stable than linear ncRNAs [101,102]. Further, circRNAs have MREs that can bind miRNAs and inactivate them, thereby increasing the expression of target genes downstream of that miRNA [103,104]. This effect is known as the miRNA sponge effect, making circRNA a ceRNA.
The subcellular distribution of circRNAs is associated with the type. ecircRNAs are mostly distributed in the cytoplasm and can serve as miRNA sponges because of the MREs they contain. Thus, they primarily exist in the cytoplasm to regulate post-transcriptional expression [105]. Correspondingly, intronic circRNAs are more often nuclear; these ciRNAs have fewer MREs and are enriched in miRNA transcription sites that regulate RNA polymerase II expression, and thus, ciRNAs primarily function at the transcriptional level [106]. circRNAs can serve as translation templates for proteins, and new evidence suggests that circRNAs can encode regulatory peptides. For example, circFNDC3B (hsa_circ_ 0006156) can encode a peptide 218 amino acids in length [107], circFBXW7 can encode a peptide 185 amino acids in length [108], and circAKT3 (hsa_circ_0017250) can encode a peptide 174 amino acids in length [109].
Involvement of circRNA in cholesteatoma development and progression and associated mechanisms
circ_0074491 and cholesteatoma of the middle ear
Using microarray and experimental data (GEO accession: GSE-102715), Hu and Qian [40] found that circ_0074491 expression was downregulated in cholesteatoma tissues. In addition, its knockdown in cholesteatoma keratinocytes promoted cell proliferation, migration, and invasion and inhibited apoptosis. circ_0074491 was shown to be a decoy for miR-22-3p and miR-125a-5p in cholesteatoma keratinocytes. Both miR-22-3p and miR-125a-5p silencing reversed these effects of circ_0074491 silencing on proliferation, apoptosis, migration, and invasion. In addition, circ_0074491 knockdown activates the PI3K/Akt pathway in cholesteatoma keratinocytes via miR-22, and circ_0074491 inactivates the PI3K/Akt pathway by binding miR-22-3p and miR-125a-5p, thus exerting an inhibitory effect in cholesteatoma. These findings provide new evidence for circRNA involvement in cholesteatoma development (Fig. 2B).
hsa_circRNA_000319, hsa_circRNA_104327, and hsa_circRNA_404655 and cholesteatoma of the middle ear
Using microarray analysis and functional prediction, Xie et al. [41] compared circRNA expression between middle ear cholesteatoma and normal skin tissues and validated differentially expressed circRNAs by qRT-PCR. In total, 13,562 expressed circRNAs were detected, with 93 upregulated (hsa_circRNA_ 104327, etc.) and 85 downregulated (hsa_circRNA_000319, hsa_circRNA_048764, etc.) compared to normal skin tissue. To validate these microarray results, eight differentially expressed circRNAs were selected for qRT-PCR analysis. Among them, there were five upregulated circRNAs (hsa_circRNA_103670, hsa_circRNA_048764, hsa_circRNA_404864, hsa_circRNA_ 104327, hsa_circRNA_404655) and three downregulated circRNAs (hsa_circRNA_101965, hsa_circRNA_000319, hsa_circRNA_100927). The validation results were partially consistent with the microarray findings, according to which hsa_circRNA_ 104327 and hsa_circRNA_404655 expression was significantly higher (P<0.05) and hsa_circRNA_000319 expression was significantly lower (P<0.05) in middle ear cholesteatoma tissue than in normal skin tissue (Fig. 2A).
According to the ceRNA hypothesis, circRNAs can bind miRNAs and repress miRNA activity through MREs. Three validated, statistically significant, differentially expressed circRNAs—namely, hsa_circRNA_000319, hsa_circRNA_104327, and hsa_circRNA_404655—were selected for a circRNA-miRNA-mRNA ceRNA network analysis, and hsa_circRNA_000319 was found to interact with hsa-miRNA-4436b-5p, hsa_circRNA_104327 was found to interact with miRNA-152-5p, and hsa_circRNA_ 404655 was found to interact with miRNA-3664-3p.
hsa-circRNA-102747 and hsa-circRNA-101458 and cholesteatoma of the middle ear
Gao et al. [42] used microarray analysis to detect circRNAs in cholesteatoma and normal skin epithelial tissues and found 13,247 circRNAs. Using the thresholds of FC>2.0 and P<0.05, 355 significantly differentially expressed circRNAs were identified. Among them, 101 circRNAs were upregulated and 254 were downregulated (FC>2.0, P<0.05). Microarray and RNA sequencing datasets were deposited in the GEO with accession number GSE102715. The circRNAs were classified as exonic (76%), antisense (3%), intronic (12%), sense-overlapping (8%), and intergenic (1%). Among the upregulated circRNAs, 79 were exonic, three were antisense, 12 were intronic, and seven were sense-overlapping, and among the downregulated circRNAs, 189 were exonic, nine were antisense, 30 were intronic, 22 were sense-overlapping, and four were intergenic.
To validate the microarray data, the researchers randomly selected six circRNAs (FC>2, P<0.05) in seven pairs of cholesteatoma and matched skin tissues for further validation. Among them, hsa-circRNA-102747 and hsa-circRNA-101458 were confirmed to be significantly different between cholesteatoma and epithelial tissues by qRT-PCR (Fig. 2C and D). Furthermore, they used GO, which provides a “framework for the model of biology” (http://www.geneontology.org) and KEGG (https://www.genome.jp/kegg/) for enrichment analysis. The GO biological functions consisted of cell morphogenesis, cell cycle, cell communication, stimulus-response, and metabolic processes. KEGG analysis revealed five significantly enriched pathways in cholesteatoma: glycosphingolipid biosynthesis, Th17 cell differentiation, galactose metabolism, Th1 and Th2 cell differentiation, and pyruvate metabolism. These were all correlated with cell growth, proliferation, migration and survival and inflammation.
Finally, the researchers used miRanda and TargetScan (http://www.targetscan.org/) for ceRNA network prediction analysis. The ceRNA network was expanded around the two previous circRNAs, 31 lncRNA nodes, 48 miRNA nodes, and 248 mRNA nodes. They hypothesized that circRNA-102747/lncRNA-uc001kfc.1/miR-21-3p/targeted mRNAs might regulate malignant characteristics, whereas circRNA-101458/miR let-7a-3p/targeted mRNAs regulate benign properties.
SUMMARY AND OUTLOOK
Current research on ncRNAs represents only the “tip of the iceberg” and is very limited, for the following reasons. First, the number of ncRNAs is much larger than that of coding RNAs, accounting for approximately 98% of all RNA. Second, ncRNAs varieties include miRNA, siRNA, piRNA, lncRNA, and circRNA, rRNA, tsRNA, snRNA, and snoRNA, but research has predominantly focused on miRNA, lncRNA, and circRNA. Third, ncRNAs are ubiquitous; they can act in the cell nucleus and cytoplasm, can be surrounded by vesicles [110–112], and play a role in exosomes. In addition, some ncRNAs can also shuttle repeatedly between the nucleus and cytoplasm, such as extra-coding RNAs (ecRNAs), which are traditionally thought to be expressed and function in the cytoplasm, and ciRNAs, which are in the nucleus. However, increasing evidence suggests that in human cancers, ecRNAs are also enriched in the nucleus and regulate transcription or splicing, such as circERBB2 [113], circHuR [114], circDONSON [115] and circDNMT1 [116]. In addition, ciRNA can also function in the cytoplasm. circAGO2 interacts with HuR to facilitate its shuttling between the nucleus and the cytoplasm [117]. Fourth, the regulation of ncRNAs is complex, involving interactions among genes, RNAs, and proteins; thus, each disease cannot be explained by a single biological factor but also cannot be separated from these interacting regulatory ncRNAs. Therefore, further research to elucidate the relevant ncRNAs will play an important role in improving disease diagnosis and treatment.
Relatively few studies currently exist on ncRNAs in middle ear cholesteatoma, with just over 10 relevant publications, but they all conclude that ncRNAs are intimately involved in the cholesteatoma formation and development. Of these, miRNA-related reports are the most numerous, with 13 (of which two also involve circRNA), followed by three circRNA-related reports (of which two also involve miRNA) and only two lncRNA-related reports. The validation of RNAs in these reports was performed by PCR. Eight studies validated their findings based on phenotypes, including proliferation, apoptosis, angiogenesis, cell cycle, invasion, migration, and inflammatory factor secretion. Five studies conducted further pathway analyses, identifying the PTEN/PI3K/AKT, angiopoietin 2, Bmi1/p-AKT, PTEN/PDCD4, NF-κB/miR-802/PTEN/p-AKT, and CDK5-related pathways as relevant. Five were selected with the intention of screening ncRNAs with large differences in expression by high-throughput sequencing or microarrays, and the remaining articles were selected by reviewing the literature to find ncRNAs with similar phenotypic effects.
These 10 publications confirm that in patients with middle ear cholesteatoma, changes in ncRNA expression levels are associated with disease progression. Among them, miRNAs were discovered and studied earliest, and thus, the findings on miRNAs are more comprehensive than those on lncRNAs and circRNAs. The mechanism underlying the effects of miRNA in middle ear cholesteatoma involves direct binding to the 3′-UTR of mRNA and regulation of the translation of proteins related to proliferation, apoptosis, invasion, metastasis, and the cell cycle, thus controlling their corresponding phenotypes. In light of the discovery of ceRNAs, miRNA research still faces many challenges. First, miRNAs can regulate the expression of several different mRNAs, and a particular mRNA can be simultaneously regulated by multiple miRNAs. Therefore, target genes cannot be sufficiently regulated by controlling the expression of only one miRNA. Second, lncRNAs and circRNAs can also regulate more than one miRNA simultaneously, and accordingly, one miRNA is also regulated by several different lncRNAs and circRNAs; thus, the regulatory network of ceRNAs is many-to-many at each node, which demonstrates the complexity of this network. Third, the proteins translated from mRNAs regulated by lncRNAs and circRNAs can, in turn, act as transcription factors to influence the transcription of lncRNAs and circRNAs. Current research on lncRNAs and circRNAs as they relate to cholesteatoma is focused on the lncRNA (circRNA)-miRNA-mRNA axis. However, lncRNAs and circRNAs usually play a regulatory role by acting as ceRNAs to prevent the binding of miRNAs and mRNAs. Compared to the simpler mechanism of miRNAs, that of lncRNAs is much more complex, and they can be involved in all forms of protein regulation. For example, lncRNAs can bind to the promoter region upstream of the coding gene and interfere with gene expression, like transcription factors [118]. They can also silence gene expression by competing with genes for transcription factors [119]. Furthermore, lncRNAs can function as scaffold-like structures linking two proteins to perform a function. They can also form complementary duplexes with transcripts of protein-coding genes, interfering with mRNA splicing to produce different splice variants [120]. Moreover, they can alter the activity or cytoplasmic localization of a specific protein by binding to that protein. lncRNAs and miRNAs can also bind and act as target molecules for miRNA and can act as precursor molecules for small RNAs, such as miRNA and piRNA [121]. lncRNAs are involved in all aspects of gene expression regulation; thus, there is considerable flexibility in the associated research, but studies on lncRNAs in cholesteatoma are currently too linear. Unlike conventional linear RNAs, circRNA molecules have a closed-loop structure and are not affected by RNA exonucleases, for which reason their expression is more stable and less susceptible to degradation. Essentially, circRNAs are classified as lncRNAs, not a new molecular type. Precisely because their characteristics are consistent with those of lncRNAs, current reports have found that circRNAs are more versatile and involved in various regulatory processes. In addition to miRNAs, circRNAs can also directly bind pri-miRNA and the promoter, coding region, or 3′-UTR of linear mRNA [122–124]. These studies have demonstrated the regulatory role of circRNAs as important RNA-binding molecules for transcription, translation, and pri-miRNA processing. However, in all relevant studies on cholesteatoma, circRNAs have only been shown to act as miRNA sponges by competitively binding miRNAs and relieving their repressive effect on target genes, resulting in upregulation of the expression of target genes. The role of other circRNAs in cholesteatoma has not been studied, and this issue requires further investigation.
In summary, studies on ncRNAs in middle ear cholesteatoma are relatively superficial, and the number of relevant papers is small. ncRNAs, in addition to the lncRNA (circRNA)-miRNA-mRNA axis, also interact with DNA and proteins to form a complex regulatory network, acting as RNA sponges, protein scaffolds, protein recruiters, enhancers of protein function, and translation templates. They are involved in transcription/splicing, translation, protein degradation, and the regulation of pri-miRNA processing. These proven roles in other diseases remain to be further investigated in cholesteatoma, thus providing new avenues for the diagnosis and treatment of middle ear cholesteatoma.
HIGHLIGHTS
▪ Cholesteatoma of the middle ear is a common disease with many related pathologies.
▪ Mammalian transcription has been found to be mainly regulated by non-coding RNAs (ncRNAs).
▪ ncRNAs play numerous and diverse roles in cholesteatoma of the middle ear.
▪ This review summarizes the roles of ncRNA types in cholesteatoma of the middle ear.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant no. 92168115 to YD); the Science Foundation for Outstanding Young Scholars of Liaoning Province (grant no. 2022-YQ-16 to YD); 345 Talent Project of Shengjing Hospital (grant no. M1400); the Project of City-University Cooperation (grant no. 2400022047 to YD); the National Key R&D Program “Active Health and Aging Technology Response” (grant no. 2020YFC2005200 to XM).
Notes
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conceptualization: all authors. Data curation: DL. Formal analysis: DL, HZ. Funding acquisition: XM, YD. Methodology: DL, HZ. Project administration: YD. Supervision: XM. Visualization: DL, YD. Writing–original draft: DL, HZ. Writing–review & editing: all authors.