Genetic Screening of GJB2 and SLC26A4 in Korean Cochlear Implantees: Experience of Soree Ear Clinic
Article information
Abstract
Objectives
Genetic hearing loss is highly heterogeneous and more than 100 genes are predicted to cause this disorder in humans. In spite of this large genetic heterogeneity, mutations in SLC26A4 and GJB2 genes are primarily responsible for the major etiologies of genetic hearing loss among Koreans. The purpose of this study is to investigate the genetic cause of deafness in Korean cochlear implantees by performing a genetic screening of the SLC26A4 and GJB2 genes.
Methods
The study cohort included 421 unrelated Korean patients with sensorineural hearing loss (SNHL) and who had received cochlear implants (CI) at Soree Ear Clinic from July 2002 to December 2010. Among 421 CI patients, we studied 230 cases who had received the genetic screening for SLC26A4 or GJB2 genes. Written informed consent was obtained from all participants. All patients had severe to profound, bilateral hearing loss. For 56 patients who showed enlarged vestibular aqueduct on their computed tomography (CT) scan, we analyzed SLC26A4. For 174 CT negative patients, GJB2 gene was sequenced.
Results
For the 56 SLC26A4 patients, 32 (57.1%) had two pathogenic recessive mutations in SLC26A4. A single recessive SLC26A4 mutation was identified in 14 patients (25%). H723R and IVS7-2A>G were the most commonly found mutations, accounting for 60.3% (47/78) and 30.8% (24/78) of the mutated alleles, respectively. For the 174 GJB2 patients, 20 patients (11.5%) had two pathogenic recessive mutations in GJB2. 235delC was the most common mutation, accounting for 43.0% (31/72) of mutant alleles.
Conclusion
The two major genes, SLC26A4 and GJB2, contribute major causes of deafness in CI patients. Continuous studies are needed to identify new genes that can cause hearing loss to Korean CI patients.
INTRODUCTION
Hearing loss is relatively common in the human population. Severe to profound sensorineural hearing loss (SNHL) is estimated to occur at approximately 1 in 1,000 births and the cause is genetic in 50% of all cases (1). Approximately 70% of congenital cases associated with genetic factors are classified as nonsyndromic. For the remaining 30%, one out of more than 400 forms of syndromic deafness can be diagnosed because of associated clinical findings. Eighty percent of cases of nonsydromic hearing loss is inherited through an autosomal recessive fashion and other cases are inherited in an autosomal dominant, X-linked, or mitochondrial pattern (2).
Genetic hearing loss is a highly heterogeneous disorder, in which a number of genes can cause the same phenotype, especially in nonsyndromic hearing loss (NSHL). Today, the chromosomal locations for NSHL are known for 50 dominant loci with 24 of the genes identified; and for 79 recessive loci with 40 of the genes found (3). In spite of this large genetic heterogeneity, mutations in SLC26A4 and GJB2 genes are responsible for major etiologies of the genetic hearing loss among Koreans (4-6).
GJB2 encodes connexin 26, a gap junction protein. Gap junctions are believed to play a role in recycling potassium ions back to the endolymph of the cochlear duct after stimulation of the sensory hair cells. The loss of connexin 26 would be expected to disrupt this potassium ion flow, thereby leading to hearing loss. Mutations in GJB2, which is located on chromosome 13q11-12, accounts for 20 to 50% of all recessive NSHL based on ethnic background (7, 8).
The second most common gene in which mutations have been reported in hearing-impaired patients is the SLC26A4 gene. Overall, SLC26A4 gene mutations associated with an enlarged vestibular aqueduct (EVA) may account for 5% to 10% of patients with prelingual hearing loss (6, 9). The SLC26A4 gene is located on chromosome 7q22-q31 and encodes a chloride-iodide transporter that is expressed in the thyroid, inner ear, and kidney. The gene has 21 exons. SLC26A4 mutations can cause both Pendred syndrome and recessive NSHL (DFNB4), two autosomal recessive disorders that share hearing loss associated with EVA or Mondini dysplasia as the common feature. The clinical distinction between DFNB4 and Pendred syndrome can be difficult to make because the goiter phenotype is variably penetrant and may not be apparent until adolescence or adult life (10).
The purpose of this study is to investigate the genetic cause of deafness in Korean cochlear implantees by performing the genetic screening of SLC26A4 and GJB2 genes. These genetic screenings can provide direct clues about the pathogenesis of hearing loss for certain cochlear implant (CI) patients.
MATERIALS AND METHODS
Recruitment of subjects
The study cohort comprised 421 unrelated Korean probands with sporadic or familial SNHL and who had received CI surgery at Soree Ear Clinic between July 2002 to December 2010. For patients with bilaterally severe to profound SNHL and without previously known causes of hearing loss (e.g., taken ototoxic drugs, or had a history of meningitis, trauma), we ordered them to screen for genetic causes before CI surgery. Except for those patients who refused to take the test, 230 patients were screened. Among 230 patients who received the genetic screening test, 210 (91.3%) were prelingual deafness. The age range of the 230 patients was 1.8 to 63 years (median age, 3 years; mean age, 8.9 years).
Preoperative auditory assessment with auditory brainstem response, or pure-tone audiometry was carried out for all CI candidates. All patients demonstrated severe to profound hearing loss. We obtained written, informed consent from adult subjects, and parents of minor subjects.
Mutation screening
We analyzed SLC26A4 for the patients who showed unilateral or bilateral EVA on their temporal bone imaging, obtained using high-resolution computed tomography or magnetic resonance imaging. EVA was defined as a diameter >1.5 mm at the midpoint of the course of the vestibular aqueduct between the posterior cranial fossa and the vestibule of the inner ear. We screened 56 patients for SLC26A4. We did not separate Pendred syndrome from DFNB4. For the other 174 patients who did not show EVA, GJB2 gene was sequenced.
Mutation screening included both exons of GJB2 (performed by sequencing the entire gene) and five hot point mutations (H723R, IVS7-2A>G, IVS9+3A>G, M147V, and 365insT) of SLC26A4 found in our subjects. We did not analyze all SLC26A4 exons in all the probands owing to limitations of DNA sample volumes and the expense of nucleotide sequencing.
RESULTS
SLC26A4 screening group
For the 56 SLC26A4 patients, 32 probands (57.1%) had two pathogenic recessive mutations in SLC26A4. Fourteen probands were homozygous for the H723R mutation, 4 probands were homozygous for the IVS7-2A>G, and 14 probands were compound heterozygotes, confirming SLC26A4-related hearing impairment. A single recessive SLC26A4 mutation was identified in 14 probands with 2 variants in SLC26A4. Five probands had no SLC26A4 mutation (Table 1).

Summary of SLC26A4 variations in Korean cochlear implantees
Among the five hot point SLC26A4 mutations, H723R and IVS7-2A>G were by far the most frequent and accounted for 60.2% (47/78) and 30.7% (24/78) of the mutated alleles, respectively. This was followed by IVS9+3A>G (2.5%), and 365insT (1.2%) (Table 2). Twelve (21.4%) of the 56 probands with EVA had an incomplete cochlear partition.
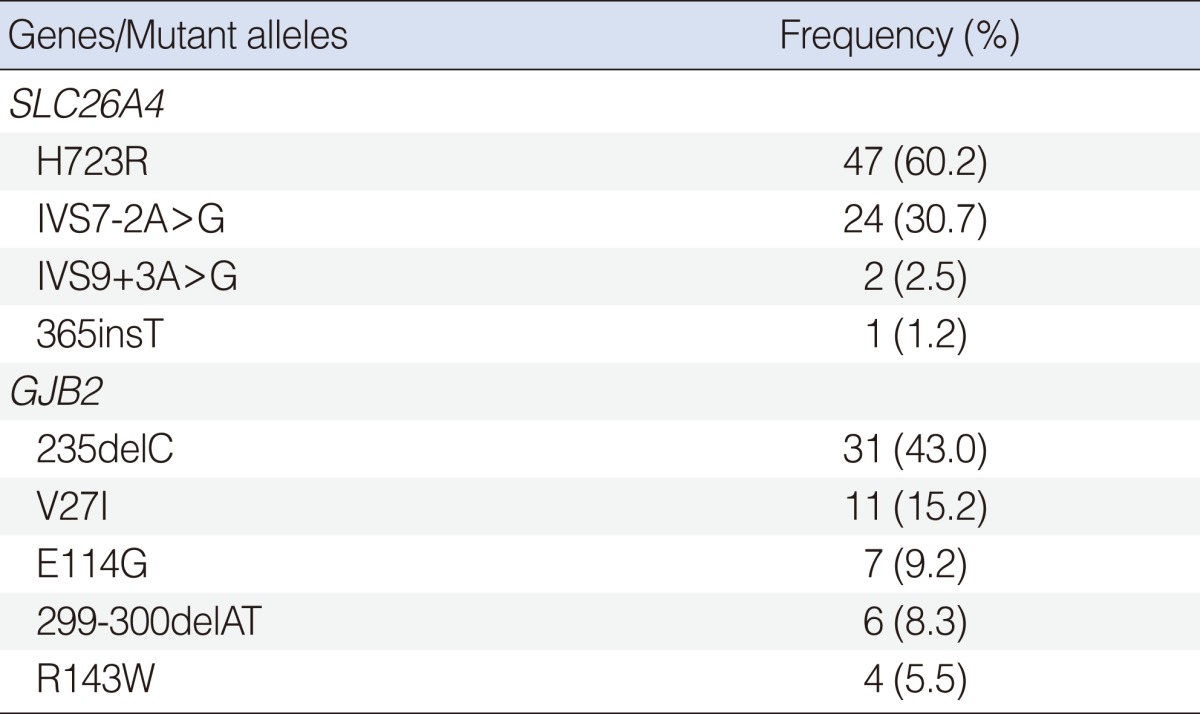
Allele frequency of SLC26A4 and GJB2 variations in Korean cochlear implantees
GJB2 screening group
For the 174 GJB2 patients, 20 probands (11.5%) had two pathogenic recessive mutations in GJB2. Gene sequencing demonstrated that 12 probands were homozygous 235delC mutation, 1 proband was homozygous R143W mutation, and 7 probands were compound heterozygote mutations. One hundred twenty-nine probands had no GJB2 mutation. A single recessive GJB2 mutation was identified in 17 probands with 7 variants (Table 3).
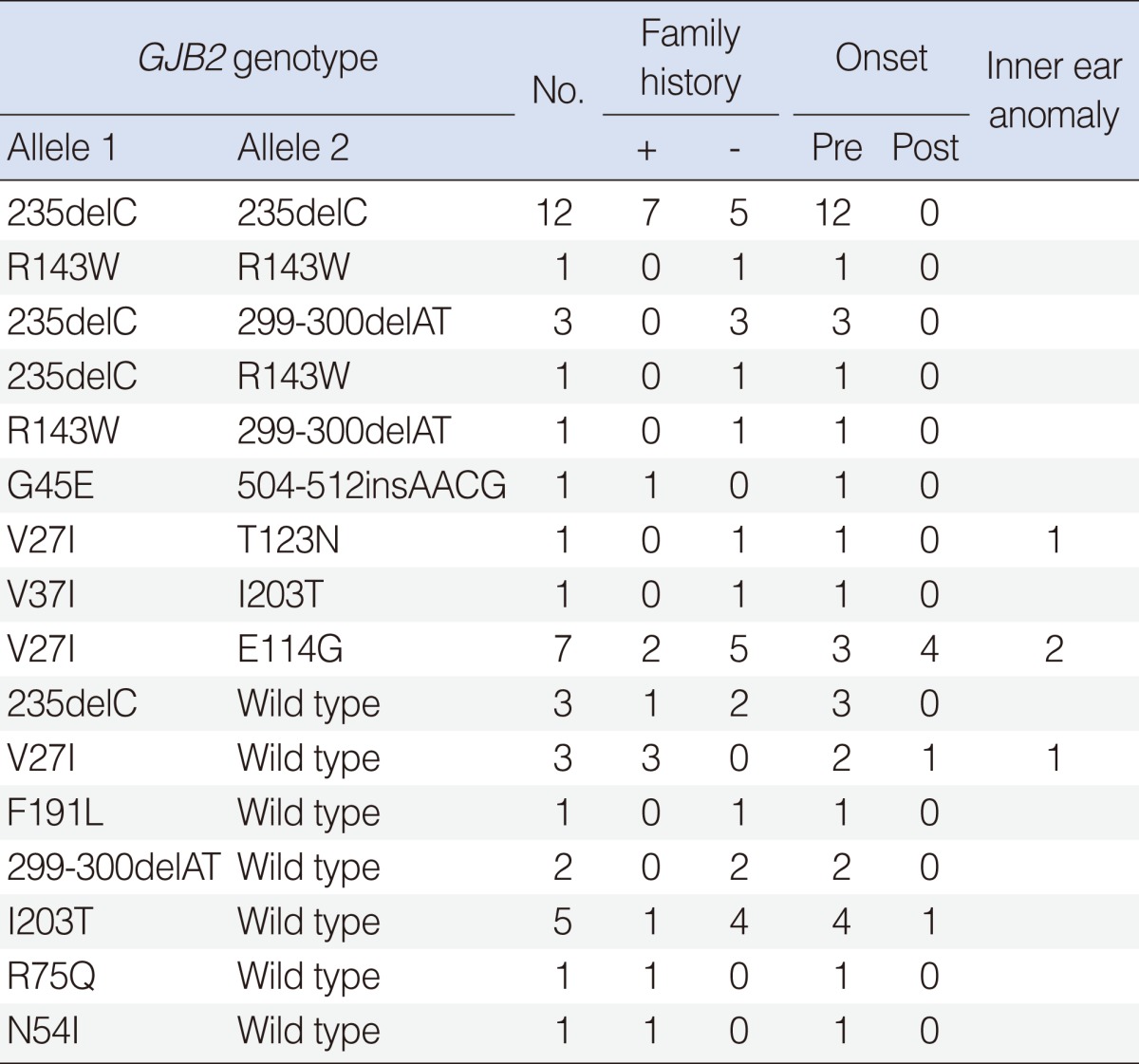
Summary of GJB2 variations in Korean cochlear implantees
The most common mutation was 235deC, accounting for 43.0% of the mutant alleles in GJB2 mutations. This was followed by V27I (15.2%), E114G (9.2%), 299-300delAT (8.3%), and R143W (5.5%) (Table 2).
DISCUSSION
Molecular genetic testing offers a new tool to determine the genetic causes of a patient's hearing loss, and its availability is rapidly growing. However, because of the extreme heterogeneity of hereditary hearing loss, screening for mutations in all genes known to be associated with hearing loss is impractical. In the present study, we performed the genetic screening of SLC26A4 and GJB2 genes, responsible for the major etiologies of hereditary hearing loss among Koreans. A definitive genetic diagnosis was made in 52 (22.6%) of the 230 CI patients, including 32 (13.9%) with two pathogenic mutated SLC26A4 alleles and 20 (8.7%) with two pathogenic mutated GJB2 alleles.
SLC26A4 mutations are causative in two autosomal recessive disorders, Pendred syndrome and DFNB4. Pendred syndrome is characterized by congenital SNHL and goiter. We did not differentiate Pendred syndrome from DFNB4 because goiter usually develops during the second decade of life and most of our cases were patients younger than 10-years-old.
According to Park et al. (5, 6), 23 (96%) of 24 probands with SLC26A4 mutations had at least one of the five recurrent mutations (H723R, IVS7-2A>G, IVS9+3A>G, M147V, or 365insT) among Koreans. As such, we screened for these five mutations.
The prevalence of SLC26A4 biallelic mutations in our series of 56 patients was 57.5% (32/56 patients) and prevalence of monoallelic mutation was 25% (14/56 patients). According to Tsukamoto et al. (11), one or two pathogenic mutations was found in 90% of Pendred families, and in 78.1% of families with DFNB4 in 32 deaf patients with EVA. Our fndings are similar.
The H723R was the most common form of mutation in 47 of 78 (60.2%) mutated alleles. The next frequent type of mutation was IVS7-2A>G, accounting for 24 of 78 (30.7%) mutated alleles. Also in the other East Asian series, H723R and IVS7-2A>G were the most common mutations (11, 12). By contrast, the mutations, IVS8+1G>A, L236P, and T416P, accounted for nearly half of all SLC26A4 mutation alleles among Caucasians (13).
Fourteen patients with EVA had one detectable SLC26A4 mutation. It is possible that these are apparent heterozygotes, or have SLC26A4 mutations that were not detected by our experimental methodology. Radiologic findings of EVA suggest it is closely related to more than one mutation of SLC26A4 (82.1%).
Up to 50% of the autosomal recessive NSHL is associated with GJB2 mutations in certain western populations (14, 15), whereas GJB2 mutations only account for 20-30% among the Japanese (16, 17). In this study, the prevalence of GJB2 biallelic mutations of 174 patients was 11.5% (20/174 patients). In addition, a previous report among Korean NSHL indicated that GJB2 mutations accounted for only 8.2% (4). These findings suggest that different types of deaf gene mutations affect variably according to ethnic background.
To date, more than 150 different variants have been reported in the GJB2 gene (18). In this study, the most common GJB2 mutation was 235delC, accounting for 31 of 72 (43.0%) mutated alleles. In other Korean and Japanese studies, 235delC was the most prevalent (4, 16, 17). The 235delC mutation results in premature termination at codon 81 and a significant loss of function of the Connexin26 protein. Many mutations are recognized to be associated with ethnic background. The 167delT is the most common connexin26 mutation among Ashkenazi Jews (19) and the R143W mutation is most prevalent in Ghana, West Africa (20). The 35delG mutation is most frequent in a majority of the Caucasian populations and may account for up to 70% of all GJB2 mutations (7, 8). However, there is no 35delG mutation in this study. Based on a previous study, there were only 2 heterozygotes among 147 Korean patients with NSHL for the 35delG mutation (4).
The next frequent variants were V27I and E114G, accounting for 12 of 73 (16.4%) and 7 of 73 (9.6%) variant alleles, respectively. The V27I and E114G variants are commonly found in the East Asian population. They are frequently found within the normal control population, and are considered a benign polymorphism (5, 17). We did not count V27I and E114G as pathologic mutations.
More than 90% of children with congenital profound hearing loss are born from parents with normal hearing, while the remaining 10% or less are born from deaf parents (21). Once a patient has been formally diagnosed with hearing loss and appropriate interventions have been implemented, clinical efforts are then directed toward finding the cause of the hearing loss. At the same time, whole genome sequencing to find the causative mutation is impractical. In this study, based on sequencing the GJB2 and SLC26A4 five hot point mutations, we were able to explain 22.6% of the causes of SNHL among deaf patients in. Identification of causative mutations can significantly help to provide adequate genetic counseling, which is primarily concerned with the planning of pregnancy and delivery with information related to a recurrence risk.
From this genetic screening, we can explain the genetic causes of SNHL to deaf patients in 22.6% of all cases (52/230), suggesting the need for a routine genetic assessment. SLC26A4 is the most frequent causative gene in Korean cochlear implant patients. GJB2 is also a second major contributor of deafness. The EVA is closely related to the mutation of SLC26A4. As our understanding of the molecular basis of hearing improves, a significant percentage of hearing loss that is presently idiopathic will likely be found to have a genetic basis. Continuous studies are needed to identify new genes that can cause hearing loss to cochlear implant patients in Korea.
Notes
No potential conflict of interest relevant to this article was reported.