Neutrophils as a Protagonist and Target in Chronic Rhinosinusitis
Article information

Abstract
Neutrophils have traditionally been acknowledged as the first immune cells that are recruited to inflamed tissues during acute inflammation. By contrast, their importance in the context of chronic inflammation has been studied in less depth. Neutrophils can be recruited and are largely present in the nasal mucosa of patients with chronic rhinosinusitis (CRS) both in Asians and in Caucasians. Increased infiltration of neutrophils in patients with CRS has been linked to poor corticosteroid response and disease prognosis. Meanwhile, tissue neutrophils may possess specific phenotypic features distinguishing them from resting blood counterparts and are endowed with particular functions, such as cytokines and chemokines production, thus may contribute to the pathogenesis of CRS. This review aims to summarize our current understanding of the pathophysiologic mechanisms of CRS, with a focus on the roles of neutrophils. We discuss recruitment, function, and regulation of neutrophils in CRS and outline the potential therapeutic strategies targeting neutrophils.
INTRODUCTION
Chronic rhinosinusitis (CRS) is a multifactorial airway disease characterized by persistent inflammation of sinonasal mucosa and associates with over 12 weeks of persistent symptoms including congestion, stuffiness, nasal discharge, pain or facial pressure, impairment or loss of sense of smell, cough, and fatigue [1,2]. It affects a considerable percentage of the population worldwide and renders substantial social and humanistic burden, and direct and indirect healthcare costs [2,3]. CRS has been thought to be a dichotomous disease according to the presence or absence of nasal polyps and are divided into two phenotypes: CRS with nasal polyps (CRSwNP) and CRS without nasal polyps (CRSsNP) [1]. Generally, CRSsNP is associated with a skewed type-1 response leading to a predominance of neutrophilic inflammation, whereas CRSwNP has a type-2 response dominated eosinophilic inflammation in Caucasian patients [4]. Patients with CRSwNP often have a higher grade of disease severity and are more likely to have a recurrent disease compared to patients with CRSsNP [5]. Recently, increasing evidences demonstrate that the inflammation patterns of CRS may vary across different geographic areas and populations with distinct racial backgrounds [4,6,7]. Significant type-2 response and eosinophilic inflammation can only be observed in 20% to 60% of patients with CRSwNP in Asia countries including China, Korea, Japan, and Malaysia [4,8,9]. In contrast, enhanced type-1 and type-17 response are found in both Asian patients with CRSsNP and CRSwNP as compared with their Caucasian counterparts [4,10]. Based on the tissue eosinophils percentage [7] or the tissue eosinophil cationic protein (ECP)/myeloperoxidase (MPO) ratio [4], CRSwNP is classified into eosinophilic and noneosinophilic type. Cao et al. [7] previously proposed a cutoff of percentage of eosinophils to classify the eosinophilic CRSwNP, which is defined when the percentage of eosinophils exceeds 10% of total infiltrating cells. This cutoff is calculated as twice of the standard deviation of the mean eosinophil percentage in control subjects [7]. Based on this classification, the authors [7] found that about half of Chinese CRSwNP patients presented noneosinophilic endotype with less impairment of olfactory function, lower frequency of asthma comorbidity, and lower ratio of ethmoid sinus/maxillary sinus CT score [11,12].
Eosinophilic and noneosinophilic CRSwNP demonstrate distinct features of epithelial cell activation, innate lymphoid cell and dendritic cell infiltration, CD4+ and CD8+ T cell subset polarization, and tissue remodeling [11,13-17]. Particularly, eosinophilic CRSwNP is consistently characterized by type-2 response, whereas noneosinophilic CRSwNP has type-1 and type17 response skewed inflammation [6,7]. Despite these differences, both subtypes of CRSwNP have been found to have upregulated neutrophil infiltration as compared to controls, and there is a higher upregulation in noneosinophilic CRSwNP than in eosinophilic CRSwNP in Chinese [6,11,16,17]. It has also been reported that CRSwNP in Caucasian patients, which is highly eosinophilic, also has significant levels of neutrophils [4,18-20]. In addition, compared to patients with eosinophilic and noneosinophilic CRSwNP, a further increase of neutrophil infiltration has been noted in patients with CRSsNP [16]. Neutrophilia affects the treatment outcome of CRS patients. It may lead to the poor response to corticosteroid therapy [21]. A recent prospective study reported by Liao et al. [22] showed that in addition to eosinophilic inflammation, significant neutrophilic inflammation was also associated with difficult-to-treat CRS in patients receiving guideline-recommended, glucocorticoid and endoscopic surgery centered treatment, underscoring a crucial role for neutrophils in the pathophysiological processes of CRS. However, compared to eosinophils, studies regarding the role of neutrophils in CRS are still in infant stage, urging the need to explore the function and regulation of neutrophilic inflammation in CRS.
This review aims to summarize our current understanding of the pathophysiologic mechanisms of CRS, with a focus on the roles of neutrophils. We discuss recruitment, function, and regulation of neutrophils in CRS and outline the potential therapeutic strategies targeting neutrophils.
LOCAL RECRUITMENT OF NEUTROPHILS IN CRS
Neutrophils are terminally differentiated granulocytes that develop in the bone marrow under the control of several key transcription factors including PU.1, CCAAT/enhancer binding protein α (C/EBP α), growth factor independence 1, and C/EBPɛ [23,24]. Following the instruction of chemokines, neutrophils rapidly leave the circulation and arrive at inflammatory sites according to the chemokine gradient by a carefully orchestrated process [25]. This process is initiated by the endothelial expression of adhesion molecules and followed by adhesion, extravasation and migration of neutrophils [23,25]. Chemokine (C-X-C motif) ligand (CXCL) family members, including CXCL1, CXCL2, CXCL5, and CXCL8, comprise the major chemokines that direct neutrophil migration through engaging G-protein-coupled receptor chemokine (C-X-C motif) receptor (CXCR) 1 and CXCR2 on neutrophils [24,26]. Both tissue infiltrating leukocytes and tissue structure cells (e.g. epithelial and endothelial cells) have been reported to produce neutrophil chemokines in response to inflammatory stimuli or foreign invaders [26]. In CRS, nasal epithelial cells are an important source of neutrophil chemokines as they have been documented to secrete CXCL8 in response to diesel exhaust particle, bacteria, or inflammatory stimuli [27-30]. In addition, CXCL1, CXCL2, and CXCL8 can be produced by fibroblasts [31,32], neutrophils [6] and mast cells [33] in nasal tissues under the stimulation with different triggers. Elevated production of CXCL8 has been found in patients with CRSsNP and CRSwNP in comparison with control subjects in China and Western countries [4,34]. Kim et al. [35] found increased expression of CXCL1, CXCL2, and CXCL8 in uncinate process mucosal samples from patients with CRSsNP compared with those from controls in Korea. Cho et al. [36] reported enhanced CXCL8 levels in nasal secretions from patients with CRSwNP compared with those from control subjects in America. Based on the unsupervised cluster analysis of protein levels of a number of cytokines and chemokines in CRS tissue lysates, Liao et al. [22] and Wei et al. [37] independently identified a neutrophil-dominated cluster associated with high levels of CXCL8 in patients in Wuhan and Sichuan city in China, respectively. These studies collectively implicate a critical role of CXCL family members in recruitment of neutrophils in CRS. However, the relevant contribution of different CXCL family members to the recruitment of neutrophils in different types of CRS need to be further clarified.
The interleukin (IL)-17 family of cytokines includes six members that play various roles in mucosal host defense and chronic inflammation [38]. Among them, IL-17A has been implicated in the recruitment and retention of mucosal neutrophils by promoting neutrophil-associated chemokine production, such as CXCL1 and CXCL8 [39,40]. Increased IL-17A levels have been demonstrated in Asian patients with CRSsNP or CRSwNP [4,6]. Wang et al. [6] found that IL-17A promoted IL-36γ production, which can further induce CXCL1, CXCL2, and CXCL8 production in CRSwNP. In addition, Wang et al. [41] found that IL-17A significantly upregulated production of serum amyloid A, which is able to promote chemotaxis of neutrophils towards epithelium in CRSwNP. However, based on cluster analysis, Liao et al. [22] and Wei et al. [37] did not find any association between IL-17A levels and the number of tissue neutrophils. Saitoh et al. [42] and Makihara et al. [43] even found that the number of IL-17Apositive cells positively correlated with the number of eosinophils, but not neutrophils, in Japanese patients with CRSwNP. These discrepancies may reflect the pleiotropic activities of IL17A in immune modulation in different inflammatory settings. For example, IL-17A has been reported to induce recruitment of eosinophils into airways in a murine model of asthma [44]. Thus more studies are needed to define the role of IL-17A in neutrophilic inflammation in CRS.
Lipid mediators are also strong inducers of neutrophil chemotaxis. Generated from arachidonic acid by 5 lipoxygenase, leukotriene B4 (LTB4), which is sensed by the LTB4 receptor on neutrophils, induces the migration of neutrophils [24]. Jeanson et al. [45] found that activation of unfolded protein response leaded to the secretion of LTB4 by nasal epithelial cells from patients with CRSwNP in vitro.
Colonized bacteria in nasal cavity in patients with CRS may induce the chemotaxis of neutrophils to local tissues. Rammal et al. [29] found that both gram-positive (Staphylococcus aureus) and gram-negative (Pseudomonas aeruginosa) bacteria stimulated CCL5 production from diseased nasal mucosal explants from CRS patients. It was also shown that N-formyl peptides, such as fMet-Leu-Phe, derived from bacterial proteins activated human neutrophils and subsequently recruited them to inflammatory sites (Fig. 1) [26,46].
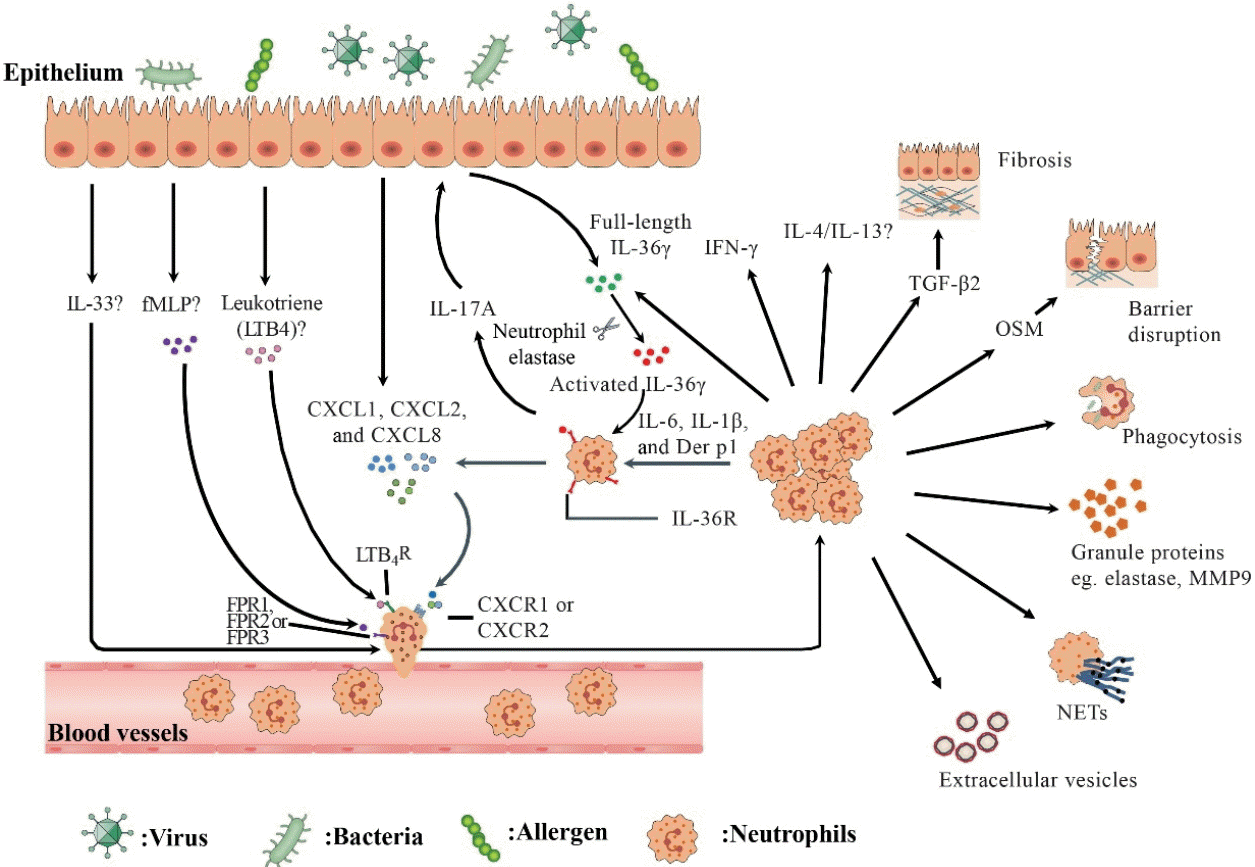
An overview of recruitment and function of neutrophils in human chronic rhinosinusitis (CRS). In response to environmental stimuli, epithelial cells secrete chemokine (C-X-C motif) ligand (CXCL) family chemokines (eg., CXCL1, CXCL2, and CXCL8) and leukotrienes to recruit neutrophils by engaging chemokine (C-X-C motif) receptor (CXCR) 1/2 and leukotriene B4 receptor (LTB4R) on neutrophils, respectively. fMetLeu-Phe (fMLP), which can be produced by colonized bacteria or damaged tissue cells, may recruit neutrophils via formyl-peptide receptor (FPR) 1/2/3. Interleukin (IL)-1 cytokine family such as IL-36γ and IL-33 participate in regulating neutrophil inflammation in CRS. IL-17A upregulates the production of full-length IL-36γ by epithelial cells. Activated IL-36γ generated by neutrophil elastase processing acts on neutrophils via IL-36R, which is upregulated by IL-6, IL-1β and Dermatophagoides pteronyssinus group (Der p) 1, and induces CXCL1, CXCL2, and CXCL8 production from neutrophils. Recruited neutrophils can kill bacteria by phagocytosis, degranulation, and formation of neutrophil extracellular traps (NETs). Activated neutrophils release extracellular vesicles. Neutrophils can also produce IL-17A, IL-36γ, IL-4/13, interferon γ (IFN-γ), oncostatin M (OSM), and transforming growth factor-β2 (TGF-β2), which may be involved in the modulation of inflammation and tissue modeling in CRS.
FUNCTION OF NEUTROPHILS IN INNATE IMMUNITY IN CRS
Currently, debate still exists on the role and profile of bacterial infection in the pathogenesis of CRS. Nevertheless, microbiota alteration has been demonstrated in patients with CRS by a number of studies [47,48]. For example, by means of conventional bacteria culture-dependent examination on nasal smear samples, Dobretsov et al. [49] reported increased colonization of S. aureus and Staphylococcus epidermidis in the nasal cavity of patients with CRSwNP compared with control subjects in western countries. Mahdavinia et al. [50] found that patients with CRS had significantly lower Corynebacterium species and Peptoniphilus species levels compared with those in control subjects by using high-throughput sequencing analysis of middle meatus swab samples. By metagenome analysis, Mahdavinia et al. [50] further found that lipopolysaccharide (LPS) biosynthesis proteins and bacterial invasion of epithelial cell pathways were significantly enhanced in patients with CRS. Bacteria prefer to exist in the form of biofilms and only 1% of all bacteria exist in the planktonic form [51]. Bacterial biofilms are a complex and organized community of bacteria encased in a self-produced exopolymeric matrix [52]. Bacterial biofilm formation may modify the inflammation process in human diseases [53]. Wang et al. [54] found that there was significantly higher number of neutrophils in biofilms-positive CRS patients compared with biofilms-negative counterparts. Neutrophils are essential for the clearance of bacteria either through phagocytosis or formation of neutrophil extracellular traps (NETs) [46].
Phagocytosis
Neutrophils are professional phagocytes. Following opsonization of the microbe and ligation of the respective opsonic receptor such as Fcγ receptors, C-type lectins, or complement receptors, neutrophils can be rapidly activated and engulf invading pathogens [46,55]. This phagocytosis process is orchestrated by a number of membrane proteins and complicated intracellular signaling cascades together with cytoskeletal rearrangements [46]. Production of oxygen radicals and their reaction products, which are collectively known as reactive oxygen species (ROS), is critical for the killing of the microbes in phagosomes [56]. However, some bacteria may impair the phagocytic capacity of neutrophils. S. aureus, which is frequently detected in Caucasian patients with CRSwNP, can inhibit phagocytosis and the subsequent killing of bacteria by neutrophils via multiple mechanisms [57]. Neutrophil phagocytosis can be regulated by toll-like receptor 2 ligand or inflammatory stimuli [58,59]. For example, pretreatment with Pam3CSK4 (a toll-like receptor 2 agonist) enhanced the phagocytic function of neutrophils against methicillin-resistant S. aureus [58]. After exposing to bronchoalveolar lavage of children with neutrophil-predominant severe asthma, neutrophils exhibited greater phagocytic capability, which is possibly a result of regulation by local inflammatory milieu [59]. However, whether the phagocytic function of neutrophils is altered in CRS remains unexplored.
Neutrophil proteases
Neutrophils have at least three types of granules including azurophilic (primary), specific (secondary), and gelatinase (tertiary) granules [60]. Azurophilic granules are consisted of MPO, neutral protease cathepsin G, elastase and proteinase 3, and defensins. Specific granules contain antimicrobial peptide lactoferrin. Gelatinase granules contain gelatinase proteins such as matrix metalloproteinase 9 [60]. During phagocytosis, azurophilic and specific granules fuse with the phagosome and release antimicrobial contents and cooperatively hydrolyze microorganisms within phagosomes, thus playing an important role in killing pathogens [25]. During degranulation or exocytosis, these preformed mediators are released from the granules into the surrounding environment [60]. Neutrophil elastase activity was found increased in nasal polyp elutes from both eosinophilic and noneosinophilic CRSwNP as compared with that in elutes from control tissues, implying a potential role of neutrophil elastase in host defense in CRS [6].
Evidence also suggests that neutrophil proteases play an important role in the coordination and escalation of inflammatory reactions through the processing of the extracellular matrix, cytokines, chemokines, and receptors [61]. For example, neutrophil elastase is able to process multiple IL-1 family cytokines, including IL-1α, IL-1β, IL-33, IL-36α, IL-36β, and IL-36γ [62]. Wang et al. [6] found that nasal polyp neutrophil elastase cleaved full-length IL-36γ into activated IL-36γ, highlighting the role of neutrophil proteases in the regulation of inflammation in CRS. Apart from elastase, neutrophil-derived MMP9 and MMP2 has been demonstrated elevated in both eosinophilic and noneosinophilic CRSwNP [6,16], although their function in CRS remains to be elucidated.
NET formation
In addition to phagocytosis and degranulation, neutrophils release their DNA in a netlike configuration to create traps known as NETs [46,63]. NETs are important for the sequestration and killing of bacteria, fungi and virus [64]. The release of nuclear content and the exteriorization of chromatin form a scaffold for the extracellular exposure of histones and antimicrobial proteins including neutrophil elastase, MPO, calprotectin, cathelicidins, and defensins [63]. NETs may kill microorganisms through the action of their antimicrobial components [64]. The formation of NETs can be triggered by microorganisms or endogenous stimuli, such as damage-associated molecular patterns and immune complexes, which has been extensively reviewed elsewhere [63]. Different functional attributes may activate different pathways to generate NETs [63]. Many stimuli, such as fungi and bacteria, activate MPO-elastase pathway leading to NETs formation. Under the stimulation, neutrophils generate ROS through the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase pathway [63]. ROS stimulates MPO to trigger the activation and translocation of elastase from azurophilic granules to the nucleus, where elastase proteolytically processes histones and disrupts chromatin packaging. Subsequently, MPO binds chromatin and synergizes with elastase to decondense chromatin, finally leading to the formation of NETs [63]. Nevertheless, some NET stimuli, such as immune complexes, ionomycin, and nicotine, may trigger NET formation independently of NADPH oxidase, relying instead on mitochondrial ROS [63]. Similar to elastase, the enzyme peptidyl arginine deiminase type 4 is critical for NET formation by regulating the initiation of DNA decondensation and nuclear envelope degradation; however, the underlying mechanisms require further investigations [46].
Currently, how neutrophils decide between phagocytosis and NET formation remains an open question. However, several studies have indicated that the pathogen attributes may be critical for this decision. Branzk et al. [65] found that neutrophils sensed the size of microbes and selectively released NETs in response to pathogens that are too large to be phagocytosed. Goodridge et al. [66] found that distinct particle-associated cellwall components, such as β-glucan, could selectively trigger a signaling cascade to initiate phagocytosis. McDonald et al. [67] found that although neutrophils were effective phagocytes by engulfing bacteria in static in vitro, upon sensing bacteria under flow condition in blood, they released their NETs into the vasculature to ensnare bacteria and to prevent dissemination.
Nevertheless, persistent NETs release can be detrimental to host. Recent studies have underscored pathogenic roles of NETs in a variety of chronic inflammatory conditions [63]. For example, elevated production of NETs has been demonstrated in the sputum of patients with cystic fibrosis and chronic obstructive pulmonary disease (COPD) and associated with disease severity [68]. Heretofore, there is few studies regarding the formation and function of NETs in CRS. Very recently, Cao et al. [69] found the formation of NETs in diseased nasal mucosa from patients with CRS. Tan et al. [70] found increased levels of autoantibodies against dsDNA, chromatin and histone in nasal polyps. It is interesting to explore whether the production of these autoantibodies is induced by relevant components in NETs in CRS in future.
Neutrophil extracellular vesicles
Extracellular vesicles (EVs) are membrane-derived vesicles that mediate intercellular communications [71]. In response to inflammatory stimuli during inflammatory processes, neutrophils also produce EVs. On the basis of the generation mechanisms, neutrophil-derived EVs can be categorized into two subtypes [72]. Neutrophil-derived trails (NDTRs) are generated during neutrophil extravasation [72], whereas neutrophil-derived microvesicles (NDMVs) are generated when neutrophils arrive at the local inflammatory sites. Although these two subtypes of neutrophil-derived EVs share some common features such as membrane components, receptors, and ligands, there are substantial differences between these two subtypes of neutrophil-derived EVs [72]. NDTRs particularly contain various chemokines including CCL2, CCL5, CXCL1, and CXCL12, which may be slowly released into the surrounding tissues [72]. NDMVs express granule-associated markers and contain granule proteins such as MPO, lactoferrin, elastase, MMP9, and proteinase 3 [72, 73]. NDTRs have been discovered in the airways of influenzainfected mice [74], and NDMVs have been found in the bronchoalveolar fluids of patients with pneumonia [75] and sepsis [76]. Recently, the functions of EVs have garnered increasing attentions. NDTRs have been shown to guide the migration of immune cells into the inflammatory sites as they release various chemokines [74], whereas NDMVs can mediate either proinflammatory or anti-inflammatory responses depending on distinct target cells [72]. For example, NDMVs promote the production of IL-6 and CXCL8 by endothelial cells, although they inhibit the production of IL-6 and IL-1β and enhance anti-inflammatory cytokine transforming growth factor-β (TGF-β) expression by natural killer (NK) cells and macrophages [72]. A recent study by Genschmer et al. [77] presents provocative new evidence that EVs released by activated neutrophils were able to bypass α1-antitrypsin, thus contributing to COPD. The authors [77] further showed that EVs were stable since EVs from COPD patients transferred the COPD phenotype to mice. However, more studies are needed to clarify the presence and stability of EVs and whether their degradation limits their pathogenic activity in pathological conditions including CRS (Fig. 1).
FUNCTION OF NEUTROPHILS IN INFLAMMATION AND TISSUE REMODELING IN CRS
Although known as innate immune cells specialized in the elimination of invading pathogens, a more complex role of neutrophils in modulating local inflammatory responses is emerging [25]. Activated neutrophils shape both innate and adaptive immune responses by producing a variety of biological mediators [78]. Peripheral blood neutrophils are known as short-lived leukocytes; however, under the control and regulation of local milieu, neutrophils in inflammatory sites may undergo profound changes with a prolonged lifespan and unique functional phenotype [63]. A recent study by Wang et al. [6] found that IL-36 receptor (IL-36R), which was undetectable on blood neutrophils, was highly expressed on the tissue neutrophils in both CRSsNP and CRSwNP. IL-36γ belongs to the recently identified IL-36 cytokine family, which is grouped into the IL-1 family. Binding to a specific IL-36 receptor complex composed of IL-36R and a co-receptor IL-1R accessory protein, IL-36γ can lead to NF-κB (nuclear factor kappa B) activation and subsequent IL-6 and CXCL8 production. The authors [6] further found that IL-36R expression on blood neutrophils could be up-regulated by IL-6, IL-1β, and Dermatophagoides pteronyssinus group 1. IL-36γ promoted the secretion of CXCL1, CXCL2, CXCL8, and IL17A from tissue neutrophils in CRS [6], suggesting that tissue neutrophils have disease-specific functional and phenotypic characteristics in CRS [6]. These findings argue for the further comprehensive comparison of tissue and peripheral neutrophils to address the regulation of neutrophils in local inflammatory site in CRS.
As one of classic Th1 cytokines, interferon γ (IFN-γ) has a significant role not only in innate immunity against pathogens but also in adaptive immunity in orchestrating leukocyte attraction, maturation, and differentiation [79]. IFN-γ was reported to induce activated but insufficient autophagy in CRS, leading to p62-dependent apoptosis of nasal epithelial cells [80]. Enhanced IFN-γ expression has been shown in both CRSsNP and CRSwNP in Asian patients, and a more predominant elevation was found in CRSsNP compared to CRSwNP [6,7]. IFN-γ can be produced by various cell types in addition to CD4+ T helper cells. Very recently, Lee et al. [81] identified that in addition to T cells, NK cells, and B cells, neutrophils also had IFN-γ expression in both CRSsNP and CRSwNP. They found that neutrophils were the predominant immune cell type having IFN-γ expression in CRS, suggesting an important function of neutrophils in inflammatory process in CRS [81].
CRSsNP, and eosinophilic and noneosinophilic CRSwNP present distinct patterns of tissue remodeling [11]. CRSsNP is highly fibrotic and CRSwNP is edematous. Nevertheless, noneosinophilic CRSwNP is less edematous and more fibrotic in comparison with eosinophilic CRSwNP [16]. Shi et al. [16] found that neutrophils were the major source of TGF-β2 in CRS. They found that the number of TGF-β2-positive cells positively correlated with the number of myofibroblasts and the expression level of fibronectin in CRS. Therefore, neutrophils may contribute to the tissue fibrosis in CRS by producing TGF-β2, particularly for CRSsNP and noneosinophilic CRSwNP.
Epithelial barrier dysfunction initiates and promotes the inflammatory and immune response in CRS [2]. Loss of barrier function makes the subepithelial components more susceptible to antigenic exposure and elicits immune response [2]. Pothoven et al. [18] found that neutrophils might disrupt nasal polyp epithelial barrier by producing oncostatin M (OSM). They demonstrated that neutrophils were a major source of OSM in CRSwNP, and granulocyte-macrophage colony stimulating factor (GM-CSF), but not IL-25, IL-33, or thymic stromal lymphopoietin, promoted neutrophil OSM production [17]. Since neutrophil itself can produce GM-CSF, Pothoven et al. [18] implied an autocrine regulatory pathway for OSM secretion in CRSwNP (Fig. 1).
Interestingly, the authors [18] showed that OSM-producing neutrophils expressed the type 2 immune response marker arginase-1, suggesting that OSM-producing neutrophils have a N2 phenotype in CRSwNP. Some studies have shown that neutrophils were able to selectively produce type 2 cytokines including IL-4, IL-5, IL-9, and IL-13 following IL-33 and IL-25 challenge [82,83], indicating a pro-allergic role of neutrophils in certain conditions. Sun et al. [82] found that IL-33-treated neutrophils displayed a distinct expression profile of cytokine and chemokine receptors as compared with resting and LPS-treated neutrophils, such as IL-13Rα1, IL-R2, and CXCR5. Similar to M1/ M2 macrophage classification, N1 and N2 neutrophil concept has also been proposed. N2 neutrophils express macrophage mannose receptor, arginase 1, chitinase-like 3, IL-10, and TGF-β and associate with tissue repair, whereas N1 neutrophils have an inflammatory phenotype expressing IL-1β and IL-12 [84]. Pothoven et al. [18] discovered that GM-CSF and follistatin-like 1 were able to induce a shifting of neutrophils from a N1 phenotype to N2 phenotype. However, the N1/N2 classification has not been widely accepted and the presence of N1 and N2 neutrophils in CRS requires further confirmation.
REGULATION OF NEUTROPHILIC INFLAMMATION BY IL-1 CYTOKINE FAMILY IN CRS
Composed of several bona fide proinflammatory cytokines including IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ, IL-1 family cytokines are instrumental as initiators and amplifiers of inflammation and are frequently among the first cytokines produced in response to infection or injury due to their constitutive expression in barrier tissues [85]. Kim et al. [86] found that the concentration of IL-1β in tissue homogenates was enhanced in noneosinophilic CRSwNP compared with eosinophilic CRSwNP and controls. Based on the unsupervised cluster analysis of protein levels of cytokines and chemokines in CRS tissue lysates, Liao et al. [22] identified a neutrophil-dominated cluster associated with high levels of IL-1β in tissue homogenates in patients in Wuhan, implicating its role in regulating neutrophilic inflammation in CRS.
The recently published study by Wang et al. [6] thoroughly explored the expression pattern of IL-36 family and its function in regulating neutrophilic inflammation in CRS. The authors [6] found that all IL-36 family members including IL-36α, IL-36β, IL-36γ, IL-36Ra, and IL-38 as well as their receptor IL-36R were upregulated in all types of CRS, eosinophilic and non-eosinophilic CRSwNP and CRSsNP, in comparison with controls. Importantly, IL-36γ was the most abundant isoform expressed and mainly produced by epithelial cells in CRS [6]. The production of IL-36γ in nasal epithelial cells was induced by double-stranded RNA, IL-1β, tumor necrosis factor-α (TNF-α), and IL-17A [6]. They further identified that neutrophils were the principal IL-36R-positive cell type in polyps and found that IL-36γ could induce the production of CXCL1, CXCL2, and CXCL8 from neutrophils in nasal polyps, which may lead to recruitment of neutrophils into nasal mucosa [6]. Interestingly, IL-36γ could also induce IL-17A production from neutrophils and later IL-17A promoted IL-36γ production from nasal epithelial cells, suggesting a positive feed-back loop of IL-17A/IL-36γ in CRS [6]. Full-length IL-36γ was produced by nasal epithelial cells in full-length form and was cleaved into active form with hundreds-fold increased biological capacity by neutrophil-derived elastase in CRS [6]. Taken together, this study implicates a critical role for IL-36γ in regulating neutrophilic inflammation in CRS.
Although IL-33 is well known for its function in initiation and exaggeration of type 2 inflammation by activating type 2 innate lymphoid cells, eosinophils, and Th2 cells [87], IL-33 may also has a role in recruiting neutrophils possibly by increasing CXCL1 production and preventing CXCR2 downregulation on neutrophils [88,89]. Kim et al. [90] found that IL-33 protein levels were elevated in noneosinophilic polyp tissues compared with eosinophilic polyp tissues. In addition, IL-33 levels were positively correlated with the levels of IL-1β, TNF-α and IL-17A, and the number of tissue neutrophils [90]. Importantly, the authors [90] discovered that anti-IL-33 treatment reduced infiltration of neutrophils, but not eosinophils, in a murine model of nasal polyps induced by ovalbumin and staphylococcal enterotoxin B, which showed a mixed type-1/2/17 inflammatory response. Kim et al.’s study [90] highlights a previously unrecognized role for IL-33 beyond type 2 response and a novel mechanism underlying neutrophilic inflammation in CRSwNP. However, the expression of IL-33 in CRS remains controversial. Liao et al. [91] found that IL-33 expression was increased in the epithelial cells in both eosinophilic and noneosinophilic CRSwNP. Baba et al. [92] found that there was no difference in IL-33 levels between polyps and controls. Thus the results regrading IL33 in CRS should be explained with caution (Fig. 1).
AGE AND NEUTROPHILIC INFLAMMATION IN CRS
Age-related differences in clinical presentation and neutrophilic immune signature has recently been noted in CRS; however, conflicting results were reported [93-95]. Kim et al. [93] reported that the number of human neutrophil elastase-positive neutrophils, myeloperoxidase gene expression, and neutrophil-associated cytokines including IL-17A and IL-23, and neutrophil chemokine receptor CXCR2 were decreased in patients older than 40 years as compared with young-aged (18–39 years) patients with noneosinophilic CRSwNP in Korean population. However, no age-related change of neutrophilic inflammation has been observed in control subjects and patients with CRSsNP or eosinophilic CRSwNP [93]. Furthermore, the authors [93] showed that elderly patients with noneosinophilic CRSwNP have better objective surgical outcomes compared with the nonelderly. In contrast, Morse et al. [94] found that although aged and younger CRS patients had similar frequencies of nasal polyps and asthma, elderly patients with CRS (≥60 years) showed elevated expression of neutrophil associated mediators in mucus including IL-1β, IL-6, CXCL8, IL-17A, and TNF-α compared with the younger counterparts (<60 years) in America. Increase in neutrophil associated mediators was linked with a higher prevalence of bacterial infection/colonization and a less likelihood to response to corticosteroids in aged patients. Cho et al. [95] found less ECP levels in nasal lavage samples in elderly subjects (60–77 years) compared with those in nonelderly subjects (16–59 years); however, there was no change of human neutrophil elastase levels and no correlation between neutrophil elastase levels and ages in patients with CRSwNP in America. These discrepancies may be due to the different techniques used to measure neutrophil-associated markers. Also, the inherited difference between the Caucasian and Asian patients could not be excluded.
THERAPEUTIC IMPLICATIONS
Eosinophilic inflammation in CRS, which is dominated by skewed type 2 response, is a relatively well-established endotype. In addition to corticosteroid therapy, biological therapies targeting IL-5, IL-4R, and immunoglobulin E are emerging for controlling eosinophilic inflammation in CRS [3]. In contrast, the neutrophilic inflammation in CRS remains very poorly defined.
Corticosteroids serve as a mainstay in CRS therapy; however, not all CRS patients respond well to corticosteroid treatment [21]. In Liao et al.’s cluster study [22], the authors identified a cluster of Chinese patients characterized by severe neutrophilic inflammation with high frequency of difficult-to-treat case despite the endoscopic sinus surgery and postoperative corticosteroid treatment, indicating a poor response of neutrophilic inflammation to corticosteroid treatment. Indeed, Wen et al. [21] found that oral prednisone was able to suppress type 2 cytokine expression and eosinophilic inflammation, whereas it could not suppress type 17 cytokine expression and neutrophilic inflammation in Chinese patients with CRSwNP. Increased neutrophilia in CRSwNP has been associated with the poor response to oral corticosteroid therapy in Chinese patients [21]. In contract to its suppressive effect on eosinophils, pharmacological studies show that corticosteroids prevent apoptosis of neutrophils and promote the neutrophilic inflammation [96].
Beyond the well-known antimicrobial effect, macrolides have intrinsic anti-inflammation and immunomodulation properties, especially for neutrophilic inflammation [97,98]. They block the activation of transcription factor nuclear factor κB and inhibit the production of various inflammatory cytokines including CXCL8 and TNF-α [97,98]. Macrolides can also suppress the secretion of airway mucus, induce the apoptosis of neutrophils, and even diminish the formation of bacterial biofilms [97,98]. Luo et al. [99] found that long-term clarithromycin treatment inhibited CXCL8 and MPO levels in Chinese patients with CRSsNP and clarithromycin was more effective for CRSsNP patients with high CXCL8 levels. However, there are conflicting results regarding the efficacy of macrolides in CRS [1,100,101]. Videler et al. [101] reported that there was a lack of efficacy of long-term, low-dose azithromycin in patients with CRS; however, the authors [101] did not performed the subgroup analysis given the insufficient study power. The value of macrolide in CRS treatment, particularly for neutrophilic inflammation, requires to be established by more well-designed studies with larger sample size.
Blocking the trafficking or activation of the neutrophils may serve as a therapeutic strategy for neutrophilic inflammation in CRS, especially for CRSsNP and noneosinophilic CRSwNP. Strategies to inhibit neutrophil recruitment may include blockage of chemokine receptors (CXCR1 and CXCR2 antagonists) and inhibition of IL-36γ, IL-17A, and LTB4. Several clinical trials assessed the effect of CXCR1 and CXCR2 antagonists and inhibitors on neutrophilic inflammation, and some have reported promising results. For example, clinical trials with a CXCR2 antagonist (MK 7123) in patients with COPD have shown that MK-7123 treatment resulted in decreased neutrophil counts and reduced MMP9 and MPO levels in plasma and sputum, and relieved disease symptoms [102]. Dual CXCR1/CXCR2 antagonist SCH527123 has been shown effective in reducing neutrophil levels in the circulation and airways in mild atopic asthmatic subjects [103]. Derived from omega-3 essential polyunsaturated fatty acids, resolvins inhibit neutrophil transmigration through downregulating neutrophil chemokines including CXCL1 and LTB4, making them a potential therapeutic candidate to suppressing neutrophilic inflammation [104]. Anti–IL-17A biologics (Brodalumab, ixekizumab, and secukinumab) have been approved by U.S. Food and Drug Administration for the treatment of psoriasis, a skin disorder characterized by enhanced neutrophil infiltration, and these agents substantially improve quality of life of patients with psoriasis [105]. However, the clinical efficacy of these agents for the treatment of CRSsNP and noneosinophilic CRSwNP remains to be studied in future.
CONCLUSION
Although increasing evidence highlight the importance of neutrophils in the pathogenesis of CRS, many aspects of neutrophilic inflammation in CRS remain unknown. These include the association between microbiome and the function of neutrophils; the difference between tissue-specific neutrophils and their blood counterparts; the heterogeneity and plasticity of neutrophils in different types of CRS; and the formation and function of NETs in CRS. Elucidating these questions will improve our understanding of the intricate role of neutrophils during the development of CRS and possibly facilitate the development of innovative therapeutics.
HIGHLIGHTS
• The inflammation patterns of chronic rhinosinusitis (CRS) vary across different geographic areas and populations with distinct racial backgrounds with more neutrophilic inflammation in Asian patients.
• Corticosteroids are the mainstay treatment for eosinophilic CRS, especially for eosinophilic CRS with nasal polyps, whereas macrolide antibiotics are possibly more effective for neutrophilic CRS.
• Targeting the activation and infiltration of neutrophils may offer potential therapeutic approaches to improve clinical outcomes in patients with steroid-insensitive CRS characterized by neutrophilic inflammation.
Notes
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conceptualization: ZL. Data curation: HW. Formal analysis: HW, LP. Funding acquisition: ZL. Methodology: HW. Project administration: ZL. Visualization: HW, ZL. Writing - original draft: HW. Writing - review & editing: all authors.
Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC; grants 81630024, 81570899, and 81325006), Natural Science Foundation of Hubei Province (grant 2017CFA 016), and “Ten thousand plan” - National high level talents special support plan of China.