Cytokines and Inflammation in Meniere Disease
Article information

Abstract
Meniere disease (MD) is a rare set of conditions associated with the accumulation of endolymph in the cochlear duct and the vestibular labyrinth with a decrease of endocochlear potential. It is considered a chronic inflammatory disorder of the inner ear with a multifactorial origin. The clinical syndrome includes several groups of patients with a core phenotype: sensorineural hearing loss, episodes of vertigo, and tinnitus with a non-predictable course. Genetic factors and the innate immune response seem to play a central role in the pathophysiology of the condition. Autoimmune MD should be diagnosed if a patient fulfills the diagnostic criteria for MD and one of the following autoimmune disorders: autoimmune thyroid disease, psoriasis, autoimmune arthritis, ankylosing spondylitis, or systemic lupus erythematosus. We summarize the evidence to support autoimmune MD as an endophenotype in bilateral MD associated with the allelic variant rs4947296 and nuclear factor-kappa B (NF-κB)-mediated inflammation, the role of cytokines (particularly interleukin-1β and tumor necrosis factor-α) in defining a subset of patients with autoinflammation, and the potential role of cytokines as biomarkers to distinguish between patients with MD and vestibular migraine. Finally, we also introduce a list of potential drugs that could regulate the immune response in MD with potential for repurposing in clinical trials.
INTRODUCTION
Meniere disease (MD) was first described by Prosper Ménière in 1861. It is a multifactorial inner ear disorder, the onset and progression of which are triggered by the combined effects of genetic, epigenetic, and environmental factors. MD is characterized by recurrent episodes of vertigo associated with ipsilateral cochlear symptoms occurring during the attacks, such as sensorineural hearing loss (SNHL), tinnitus, or aural fullness [1,2]. Most patients progress to chronic imbalance, moderate to severe deafness in the affected ear and, in many cases, persistent and disabling tinnitus [3]. The disorder usually begins in one ear with tinnitus and hearing loss (unilateral MD; UMD), but it can evolve to both ears and produce bilateral symptoms (bilateral MD; BMD). The diagnostic criteria for MD are based on the joint presentation of vertigo and aural symptoms during the attack, and they were reformulated jointly by the Classification Committee of the Bárány Society, the Japan Society for Equilibrium Research, the European Academy of Otology and Neurotology, the Equilibrium Committee of the American Academy of Otolaryngology-Head and Neck Surgery, and the Korean Balance Society in 2015 [4]. However, these criteria do not consider the clinical heterogeneity observed among patients, and some comorbidities commonly observed in some patients, such as migraine or autoimmune disease (AD), should be considered in the therapeutic management.
CLINICAL SUBGROUPS IN MD
MD is not a single disease; instead, it should be considered as a clinical syndrome with different etiologies. Ten clinical subgroups of MD patients have been described using cluster analysis and clinical data from over 1,500 MD patients [5,6], according to four clinical predictors: cochlea-vestibular symptoms occurring after SNHL (delayed hydrops), familial MD (FMD), migraine, and AD. Five subgroups were reported in UMD and five were also found in BMD (Fig. 1, Table 1), all with strong potential etiological implications, and six of these subgroups were shared between UMD and BMD (type 3, FMD; type 4, MD with migraine; and type 5, MD with AD).
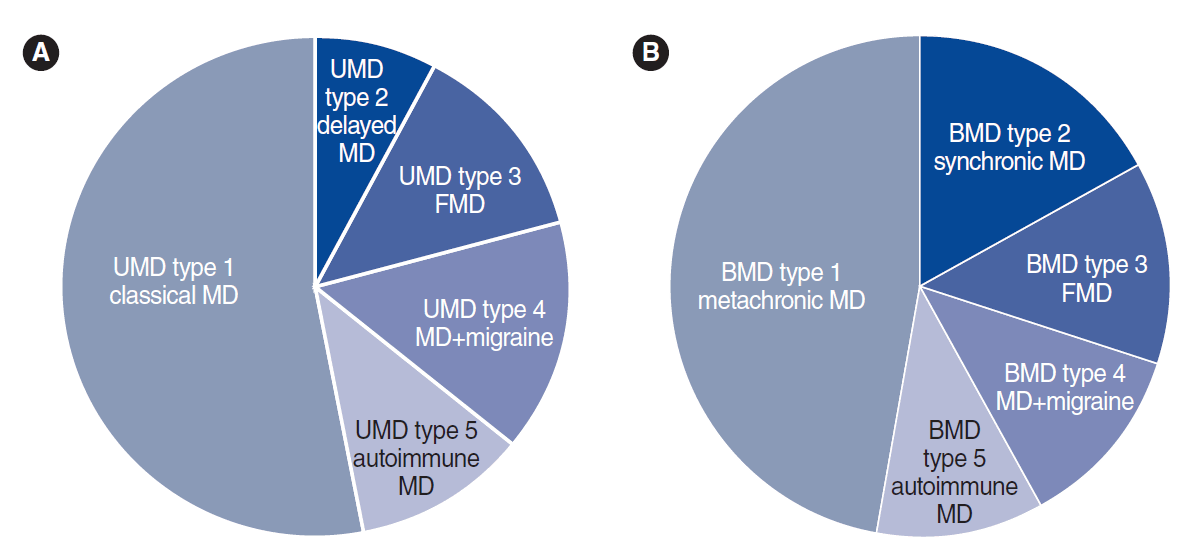
Clinical subgroups of patients with unilateral Meniere disease (UMD; A) and bilateral Meniere disease (BMD; B). MD, Meniere disease; FMD, familial Meniere disease.
Clinical subgroups of patients with UMD and BMD
Familial clustering has been reported in about 9% of cases in southern European populations [7], and in 6% of cases in South Korea [8], additionally supporting a genetic contribution to the disease [9]. FMD presents an autosomal dominant pattern of inheritance with incomplete penetrance and anticipation, showing an earlier onset compared to sporadic MD [10-12].The families can include patients with UMD or/and BMD; therefore, epigenetic factors might influence unilateral or bilateral involvement [7]. FMD could be split into two subgroups (FMD with and without migraine), confirming the early description of families with MD co-segregating with migraine [13] and the more recent description of FMD without migraine [14,15], as well as reflecting the genetic heterogeneity of FMD. Familial cases may also show recessive inheritance, providing further support that several genes contribute to FMD and suggesting that it should be considered a rare polygenic disease [16].
These clinical variants observed in both UMD and BMD validate previous epidemiological studies, but they also indicate a separate role for genetics and autoimmunity as relevant factors influencing the development of MD. While the pathological mechanisms underlying MD remain poorly understood, many studies have postulated that various factors may be involved, including endolymphatic hydrops, allergy, inflammation, infection, and ADs [17,18]. Among these theories, it seems that the immune system plays a critical role in a large population of MD patients [19,20]. However, the role of the immune system in patients with MD has not been well studied.
THEORY OF IMMUNE INVOLVEMENT IN THE INNER EAR
The notion that the immune system could have a role in some idiopathic hearing loss and vestibular disorders was introduced during the early 20th century by Joannovic in 1920 [21] and Masugi in 1931 [22]. In 1958, Lehnhardt [23] suspected that certain cases of sudden bilateral hearing loss could be associated with the production of anti-cochlear antibodies. Kikuchi [24] suggested an autoimmune etiology after observing that surgery in one ear affected the other one. Beickert [25] and Yoshihiko and Yukihiro [26] both presented data supporting an autoimmune mechanism in experimental guinea pig cochleae. In 1979, McCabe [27] first described patients with bilateral progressive hearing loss that responded to steroid therapy. The clinical presentation of SNHL can be quite variable, often overlapping with other disorders such as MD or deafness autosomal dominant 9 (DFNA9), which can result in diagnostic confusion. DFNA9 is caused by pathogenic variants in the COCH gene, which encodes cochlin [28]. Cochlin, a major component of the extracellular matrix in both the cochlea and vestibule of the inner ear, is a potential candidate antigen for autoimmune inner ear disease (AIED) [29] and DFNA9 with or without vestibular abnormalities [30], related to inner ear immunity and stimulation of the secretion of inflammatory cytokines [31]. Hughes et al. [32] reported that over 52% of patients diagnosed with AIED presented hearing loss and vertigo, suggesting that a continuum might exist between MD and SNHL (Fig. 2).
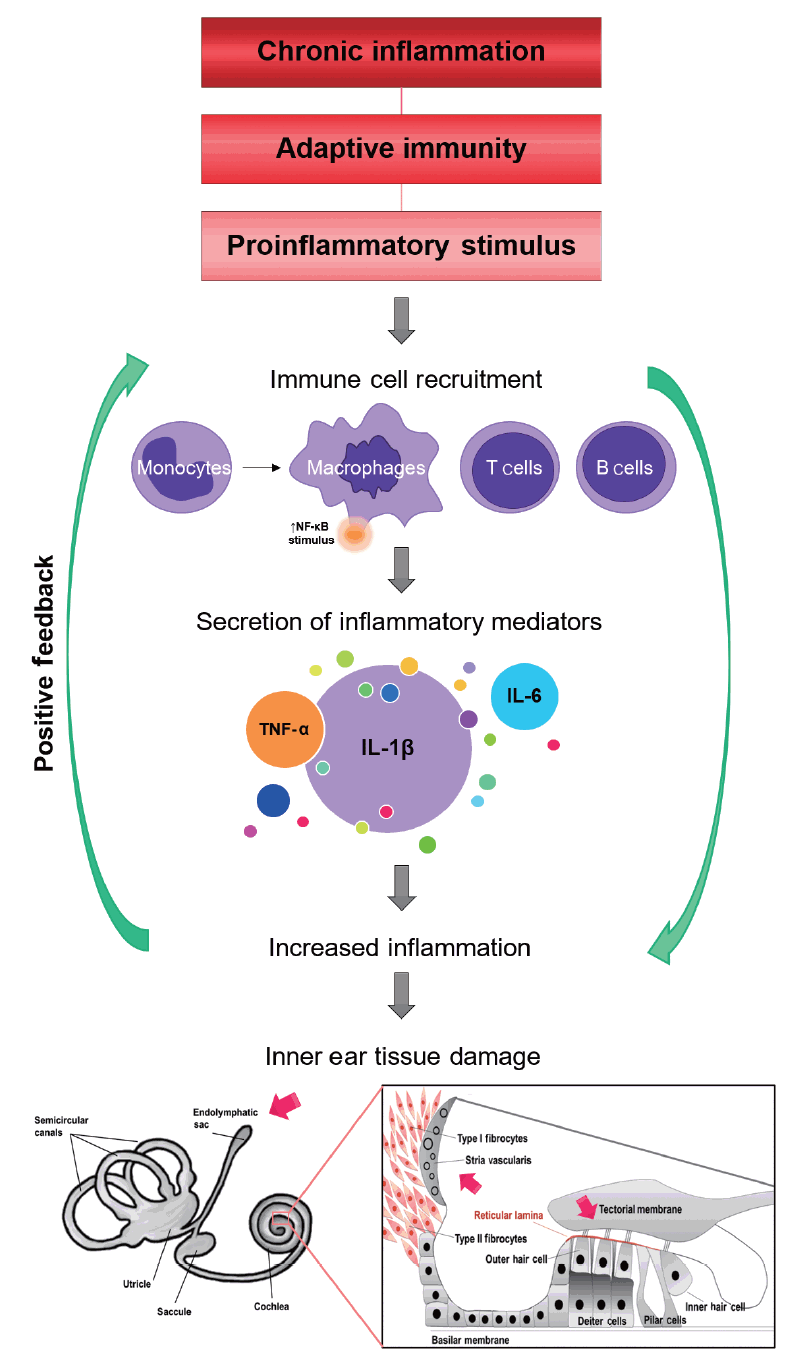
Potential mechanisms of inflammation in Meniere disease (MD). Red arrows indicate the potential targets most likely to be damaged. NF-κB, nuclear factor-kappa B; TNF, tumor necrosis factor; IL, interleukin.
AUTOIMMUNE MD
We currently have evidence of inflammation in some inner ear diseases, including MD, progressive SNHL, otosclerosis, and sudden deafness. The prevalence of systemic ADs such as rheumatoid arthritis (RA), ankylosing spondylitis, systemic lupus erythematosus (SLE), and psoriasis in MD patients is 3- to 8-fold higher than in the general population [33-35]. According to some studies, autoimmunity seems to be responsible for 6% of cases of UMD and 16% of cases of BMD [36]. Based on the findings of proteomic studies performed in small series of patients, autoimmunity has been proposed as a potential cause of MD [20,37]. Approximately one-third of MD cases seem to be of autoimmune origin; however, elevated immune complexes were only found in 7% of patients with MD [38], and there is no consistent immunological biomarker for the diagnosis of MD. Therefore, several hypotheses have been proposed to explain how inflammation may arise in MD: bystander damage, cross-reactions, intolerance, and genetic factors. These mechanisms are supported by several experimental studies, as detailed below. (1) Antibodies or rogue T cells may cause accidental inner ear damage because the ear shares common antigens with a potentially harmful substance, virus, fungus, or bacterium that the body is battling. This is presently the preferred theory for AIED. (2) The body may not recognize all inner ear antigens. When they are released (perhaps after surgery or an infection), the body may wrongly attack the “foreign” antigens. In the ear, a mechanism that could be involved in so-called sympathetic cochleo-labyrinthitis has been reproduced in animal models [39]. (3) Endolymphatic hydrops can be induced experimentally by injection of antigens or monoclonal antibodies [40]. Inner ear antigens with molecular weights of 28, 42, 58, and 68 kDa could be the main components that induce autoimmune MD in guinea pigs [41]. (4) The deposition of circulating immune complexes (CIC) could produce inflammation and interfere with the capability of endolymphatic sac (ES) filtering. Several studies have demonstrated increased CIC levels in 21%–96% of MD patients [42]. (5) Autoantibodies have been found in MD patients’ sera [43]. (6) Genetically controlled features of the immune system could increase or otherwise be associated with increased susceptibility to common hearing disorders [44]. Allelic variants in immune response genes related to SNHL progression, such as MICA, TLR10, and NFKB1, have also been reported [45-47]. (7) Frejo et al. [48] have shown that an immune response factor involved in MD is Fn14 (fibroblast growth factor-inducible 14; Tnfrsf12a), the receptor for TWEAK, a multifunctional cytokine (tumor necrosis factor [TNF]-like weak inducer of apoptosis; Tnfsf12), which is a member of the TNF superfamily. The TWEAK/Fn14 pathway is involved in the modulation of inflammation in several chronic ADs, including multiple sclerosis (MS), SLE, RA, and ulcerative colitis [49]. It has been discovered that one of its allelic variants (rs4947296) is a quantitative trait locus that regulates the expression of multiple genes in the TWEAK/Fn14 pathway in peripheral blood mononuclear cells. This locus leads to an inflammatory response mediated by the transcription factor nuclear factor-kappa B (NF-κB) in MD. This variant was also associated with bilateral MD in the Spanish population, and it appears in up to 18% of patients with comorbid autoimmune conditions.
However, the evidence supporting the hypothesis of autoimmunity is limited and the involved immunological mechanisms remain unclear. Autoimmune MD should be diagnosed if a patient fulfills the diagnostic criteria for MD and one of the following ADs: autoimmune thyroid disease, psoriasis, autoimmune arthritis, ankylosing spondylitis, or SLE. All of these conditions have been previously associated with MD in epidemiological studies in European populations [33,35].
CYTOKINES IN THE INNER EAR
The existence of immunological activity in the anterior labyrinth has been widely described, both in humans and animal models. Immune responsiveness in the inner ear was initially associated with the ES [50-52] since it possesses immunological capacities and is responsible for a major part of the trans-epithelial ion transport occurring within the inner ear [53]. However, the presence of immune capacity in the cochlea has since been established [54] through the recent demonstration of IBA1-expressing macrophages [55,56] in the human ES and cochlea that express major histocompatibility complex type II (MHCII). Kampfe Nordstrom et al. [57] identified that the macrophage population of the stria vascularis, spiral ligament, and spiral ganglion expressed MHCII, which is crucial for initiating antigen-specific immune responses. Furthermore, it was proposed that there could be uptake and processing of antigens from the ES lumen, due to the co-expression of IBA1 and MHCII in epithelial cells and transepithelial migration. Additionally, Moller et al. [58] found gene expression for both the cellular and humoral innate immune-system, including toll-like receptors 4 and 7, beta-defensin, and lactoferrin in the ES. These findings provide molecular evidence of an immunological capacity of the ES to recognize and process antigens for immune responses.
Autoinflammatory diseases are triggered by an overactive inflammatory response leading to immune dysregulation, fundamentally mediated by interleukin (IL)-1β, type I interferon (IFN)-mediated responses, and the transcription factor NF-κB [59]. These diseases normally manifest in the perinatal period; however, late-onset forms are diagnosed in adulthood [60]. Some of these disorders have symptoms that mimic allergic and immunodeficiency disorders. Additionally, many of them are accompanied by SNHL as a symptom, suggesting that potentially similar hearing loss pathogenesis molecular mechanisms may exist. Over the last decade, new molecules have been explored to detect the role of cytokines in AD, and new drugs are being developed to interfere with them. Around 60% of MD patients have antibodies in their sera against proteins in the inner ear, and there is evidence of the presence of cytokines in the cochlea, including IL-1α, TNF-α, NF-kβ P65, P50, and Ikβα [61].
TNF-α
TNF-α is a cytokine that binds to TNFRSF1A/TNFR1 and TNFRSF1B/TNFBR. It is mainly secreted by macrophages, induces the infiltration of immunocompetent cells into the tissues, and amplifies the immune response. It is a potent pyrogen causing fever by direct action or by stimulation of IL-1 secretion, and under certain conditions it can stimulate cell proliferation and induce cell differentiation. It may also impair regulatory T-cell (Treg) function in individuals with RA via FOXP3 dephosphorylation. TNF-α upregulates the expression of protein phosphatase 1, which dephosphorylates the key Ser418 residue of FOXP3, thereby inactivating FOXP3 and rendering Treg cells functionally defective [62].
In a small study population of 15 subjects, Ren et al. [63] reported that both TNF-α and IL-6 levels were significantly elevated in patients with sudden SNHL and progressive SNHL when compared to controls. This study was followed by a preliminary report on the use of etanercept [64], a well-known TNF-α blocker used in ADs such as psoriasis and RA, for patients with immune-mediated cochleovestibular disorders who did not respond to conventional therapies. They reported a 92% success rate in hearing loss improvement with etanercept therapy.
Animal models of labyrinthitis have been used to study the role of TNF-α in recruiting inflammatory cells to the cochlea and its role in resultant hearing loss [65]. While some researchers reported that, in an animal model, etanercept had a protective effect on hearing loss [66], others found it to be less effective than prednisone, with a greater potential for adverse effects [67]. In contrast, human studies performed in patients with sudden SNHL showed increased levels of TNF-α [68], while other studies presented opposite results [69]. In 2012, Svrakic et al. [70], performed a prospective study in patients with immune-mediated SNHL (IM-SNHL) where they concluded that TNF-α had the potential for use as both a diagnostic marker for IM-SNHL and a prognostic biomarker for corticosteroid response in the disease. Three years later, Pathak et al. [71] demonstrated the TNF-α could be attenuated in a subset of patients with AIED using N-acetylcysteine (NAC) [72], which might serve as a beneficial adjunctive therapy in hearing restoration. NAC has been shown to have a protective adjunct role in idiopathic sudden hearing loss, as the addition of NAC to corticosteroid therapy resulted in better hearing recovery than with corticosteroids alone [73].
IL-1β
IL-1β is produced by monocytes as an inactive 31 kDa precursor, termed pro-IL-1β, in response to molecular motifs carried by pathogens referred to as pathogen-associated molecular patterns (PAMPs). Pro-IL-1β is cleaved by the protease caspase-1 [74]. The activation of caspase-1 occurs via recruitment to a multi-protein complex termed the inflammasome [75], which leads to an active 17-kDa form of IL-1β [76].
Over a decade ago, the critical role of the IL-1 family as regulators of inflammation and immunity became evident [77]. Early immune system reactions to PAMPs may drive many of the later adaptive T cell responses that maintain the disease. IL-1β is a key proinflammatory cytokine involved in the progression of many autoinflammatory diseases and ADs, including AIED and MD [78]. During the immune response, the absence of the expression of IL-1 receptor antagonist (IL-1Ra) or other molecules that oppose the IL-1β inflammatory cascade could promote the development of ADs and autoinflammatory diseases. IL-1β is crucial in the development of inflammation and the aggravation of many autoinflammatory diseases and ADs. IL-1β plays a homeostatic role in routine biological processes, but its constitutive overproduction is associated with chronic diseases such as RA, osteoarthritis, type 1 diabetes, MS, inflammatory bowel disease, Muckle-Wells syndrome, DFNA34, and corticosteroid-resistant AIED [79-87]. IL-1β is processed by the inflammasome, and genetic mutations occurring in its different components and the resultant overproduction of IL-1β [88,89] are clinically manifest in many autoinflammatory diseases [90-92]. The role of IL-1β in hearing disorders is essentially unknown; nonetheless, there are examples of its involvement in both animal models of AIED and clinical autoinflammatory disorders with associated SNHL [93]. SNHL has been observed as a component of clinical diseases involving IL-1β dysregulation, such as neonatal-onset multisystemic inflammatory disease syndrome [94] and Muckle-Wells syndrome [95]. Likewise, improvement of SNHL has been observed in response to treatment with anakinra, a soluble IL-1Ra, in Muckle-Wells syndrome and AIED [96,97]. A recent report found that the processed 28-kDa form of IL-1β, which is uniquely generated by caspase-7, in patients with AIED can activate further downstream proinflammatory cytokines [78].
CYTOKINES AND MD
Frejo et al. [98] observed that basal levels of the proinflammatory cytokines TNF-α, IL-1β, and IL-6 could be increased in some MD patients. This observation was found in both UMD and BMD patients, as well as in healthy individuals after stimulation with lipopolysaccharide (LPS), which was used as positive control for inflammation (Fig. 3). They observed two subgroups of MD patients according to their IL-1β profile (patients with high levels of IL-1β and patients with low levels of IL-1β), and these groups could have different immune responses or functional states of the immune system [98]. Later, Flook et al. [99], showed that patients with MD or vestibular migraine had separate pro-inflammatory profiles. A cytokine panel including IL-1β, CCL3, CCL22, and CXCL1 could be used as biological markers for the differential diagnosis of vestibular migraine and MD. The expression of IL-1β, CCL3, CCL22, and CXCL1 has been previously observed in mouse cochlear tissue [100], and IL-1β, CCL3, and CXCL1 have also been found in cells from the ES [101]. Interestingly, CXCL1 is the only chemokine that has been found to be expressed in mouse vestibular cells [102], and it showed the highest expression of the four cytokines in all tissues. Therefore, cytokine and chemokine profiles could be used to define the immune response and the functional state of the immune response in MD.

Temporal profile of cytokine release after lipopolysaccharide (LPS) stimulation in peripheral blood mononuclear cells from a healthy individual. IL, interleukin; TNF, tumor necrosis factor.
POTENTIAL DRUGS THAT COULD REGULATE THE IMMUNE RESPONSE IN MD
The treatment of MD primarily focuses either on lowering endolymphatic pressure or controlling its symptoms during vertigo episodes. No treatment cures the disease; therefore, treatment involves a trade-off of improving symptoms with the fewest side effects. Consequently, treatment starts with the most conventional modalities: diet, lifestyle changes to reduce stress, and medications to control vertigo. Hence, better treatment options are needed. Implicitly, all clinically employed drugs have effects on biological systems other than those for which they were designed. This characteristic of drugs could result in a positive outcome, as occurs when a drug established for one therapeutic indication finds utility for another indication (known as “repurposing”). For example, steroids, which are commonly used in ADs, can benefit patients with sudden hearing loss or autoimmune MD (Table 2).

Potential beneficial drugs for the treatment of autoimmune/autoinflammatory Meniere disease
Several actors including immune and non-immune cells, cytokines such as TNF-α, IL-1β, IL-6, or type I IFN, and transcription factor NF-κB play central roles in inflammation. Synergistic interactions between NF-κB and other transcription factors induce the hyper-activation of NF-κB, followed by the production of various inflammatory cytokines [103]. TWEAK is a multifunctional cytokine that regulates several cellular responses, including inflammation, cellular adhesion, proliferation, and apoptosis [104,105]. TWEAK is expressed in several cell types, including monocytes/macrophages, dendritic cells, activated T cells, astrocytes and microglia, endothelial cells, and erythroblasts. The biological activity of TWEAK is mediated through its receptor, fibroblast growth factor-inducible 14 (Fn14), which is highly expressed in epithelial cells and induced in several human diseases [106]. Increased levels of TWEAK and/or Fn14 have also been found to be associated with the pathogenesis of RA [107], SLE [108], MS [109], or neuroinflammation [110]. The binding of TWEAK to Fn14 induces both acute activation of the canonical NF-κB pathway and prolonged activation of the non-canonical NF-κB pathway [104]. Furthermore, the non-canonical NF-κB pathway plays a key role in immunity and immune-mediated disorders such as SLE. The non-canonical NF-κB pathway relies on the phosphorylation-induced p100 processing, which is triggered by signaling from a subset of TNFR members, including Fn14, TNFR2, BAFFR, CD40, LTβR, and RANK [111]. Most of these signals are regulatory elements of the immune response, supporting the hypothesis that the allelic variants of immune response genes can modify the clinical course in MD. Inflammation may occur in the ES, since proteomic studies have found high contents of immunoglobulins (Igs) in the ES [20]. Endolymph and perilymph homeostasis is maintained at the cochlea and vestibular semicircular canals at multiple sites, including the spiral ligament, the stria vascularis, the cochlear and vestibular non-sensory epithelial cells, and the ES. The sac is a small organ located in the posterior cranial fossa and plays a crucial role both in the maintenance of endolymph composition and in the innate immune response [58]. We hypothesize that, after exposure to an environmental trigger, the carriers of the risk haplotype could have an abnormal NF-κB-mediated inflammatory response in the ES, causing an ionic imbalance in the endolymph, leading to the accumulation of endolymph at the cochlear duct. Therefore, the TWEAK/Fn14 and NF-κB pathways have potential druggable targets.
Although there are no approved drugs for either TWEAK or its receptor Fn14, BIIB023 is an anti-TWEAK monoclonal antibody that demonstrated a favorable safety and tolerability profile in a phase 1 study of RA patients. In contrast, regulation of NF-κB pathways play a pivotal role in cellular responses to changes in the environment. At present, several biologics and molecules that modulate both the canonical and non-canonical pathways are in the market or in clinical trials. Moreover, many marketed drugs that were later observed to also have NF-κB targeting activity were repurposed for new therapeutic interventions [112]. There are over 700 known inhibitors of NF-κB signaling at several levels [113] that could be potential druggable targets. The NF-κB pathway in general can be regulated at three different points: (1) blocking the signal reception by interfering with the receptor-ligand binding, thereby abolishing the signaling cascade at the initial stage; (2) interfering with any of the factors involved in the cascade in the cytoplasm; and (3) targeting the nuclear translocation of NF-κB factors or interfering with their DNA binding.
Cepharanthine (CEP) has a multifactorial mechanism of action. The drug exerts membrane effects such as the modulation of efflux pumps and membrane rigidification, as well as various intracellular and nuclear effects. CEP interferes with numerous metabolic axes, primarily with the AMP-activated protein kinase (AMPK) and NF-κB signaling pathways. Specifically, its anti-inflammatory effects rely on AMPK activation and NF-κB inhibition. Resveratrol inhibits NF-kB signaling through suppression of p65 and IκB kinase activities [114].
Several cytokines could also be potential targets to treat MD since they mediate an overactive inflammatory response that triggers immune dysregulation in inflammatory diseases. Type I IFN can have dual and opposing roles in immunity, with effects that can be beneficial or detrimental to the individual depending on whether IFN pathway activation is temporary or persistent. When dysregulated, the type I IFN system drives many inflammatory diseases, including SLE, myositis, RA, and Sjogren syndrome [115]. IL-6 is a pleiotropic pro-inflammatory cytokine produced by a variety of cell types, including monocytes, lymphocytes T- and B-cells, and fibroblasts. IL-6 has been shown to be involved in diverse physiological processes in response to infections and tissue damage, and it contributes to host defense through the stimulation of acute-phase responses, hematopoiesis, and immune reactions. While its expression is firmly regulated by transcriptional and posttranscriptional mechanisms, dysregulated continual synthesis of IL-6 exerts a pathological effect on chronic inflammation and autoimmunity [116].
There are several approved drugs against IL-6 and its receptors (IL-6R and IL-6ST). Siltuximab is a recombinant human-mouse chimeric monoclonal antibody that binds to IL-6. It prevents the binding of IL-6 to both soluble and membrane-bound IL-6 receptors (soluble IL-6R [sIL-6R] and membrane IL-6R [mIL-6R]), inhibiting IL-6 signaling. Sarilumab is a human recombinant monoclonal antibody of the IgG1 subclass. It binds to both sIL-6R and mIL-6R, and it has been shown to inhibit IL-6-mediated signaling through these receptors. Tocilizumab is a first-generation antibody directed against IL-6R. It inhibits binding between IL-6 and IL-6R. Satralizumab is a second-generation anti-IL-6R monoclonal antibody designed to have enhanced antigen-neutralizing capacity and a longer plasma half-life than tocilizumab. Furthermore, several anti-IL6 agents, such as clazakizumab, and anti-IL6R agents, such as olokizumab and vobarilizumab, are also being studied.
TNF has two forms: soluble (solTNF) and transmembrane (tmTNF). Although both forms are biologically active, they have clearly different roles. tmTNF has been shown to play a crucial role in maintaining the physiological innate immune response to infections [117], while the main role of solTNF is to drive the inflammatory response. This function has been proven in animal studies showing that inhibition of solTNF causes an anti-inflammatory effect. Inhibition of tmTNF, in contrast, results in increased sensitivity to infection and exacerbation of demyelination. Five TNF inhibitors have been approved for the treatment of various inflammatory diseases: etanercept, infliximab, adalimumab, certolizumab, and golimumab. All of these agents competitively inhibit the binding of TNF to its receptors. Nevertheless, they differ in both pharmacokinetic and pharmacodynamic characteristics, leading to significant differences in their clinical efficacy and indications. Alongside the approved therapies (RA, ankylosing spondylitis, Crohn disease, and psoriasis), TNF inhibitors are also repurposed for off-label indications such as SLE, scleroderma, Sweet syndrome, or Sjogren syndrome, even though in most of these cases large, controlled studies are still lacking [118].
Adalimumab is a humanized monoclonal antibody that, on one hand, specifically binds to TNF and blocks its interaction with endogenous TNF receptors to modulate inflammation, while on the other hand, it induces apoptosis of lymphocytes that are abnormally activated. Certolizumab is a polyethylene-glycolated Fab’ fragment of TNF antibody that specifically binds to TNF-α and neutralizes it in a dose-dependent manner. It also inhibits the production of LPS-induced TNF-α and IL-1β. Golimumab is a human monoclonal antibody with immunosuppressive features that targets both solTNF-α and tmTNF-α. The TNF-golimumab complex neutralizes the expression of C-reactive protein levels, IL-6, intercellular adhesion molecule-1, matrix metalloproteinase-3, and vascular endothelial growth factor induced by TNF-α, demonstrating that golimumab is an effective modulator of inflammatory markers [119]. Etanercept is a recombinant version of soluble human TNF receptor fused to an IgG Fc fragment that binds specifically to TNF and inhibits its binding with endogenous TNF receptors. The products of infliximab neutralize the biological activity of TNF-α by binding with high affinity to both solTNF-α and tmTNF-α, inhibiting their binding with TNF-α receptors. Infliximab products do not neutralize TNF-β (lymphotoxin-alpha), a related cytokine that employs the same receptors as TNF-α.
More than any other cytokine family, the IL-1 family is primarily associated with innate immunity, and even though the inflammatory characteristics of the IL-1 family dominate in the innate immune response, they can also play a role in acquired immunity [120]. IL-1β is a proinflammatory cytokine that exerts pleiotropic effects on a variety of cells and plays key roles in acute and chronic inflammatory disorders and ADs. Its overproduction is implicated in the pathophysiological changes that arise during different disease states, such as RA, MS, Alzheimer disease, neuropathic pain, and inflammatory bowel disease. The proinflammatory activities of IL-1 are controlled by various endogenous inhibitors, such as IL-1Ras and membrane-bound and soluble IL-1 receptor type II and IL-1 receptor accessory proteins [121,122].
Canakinumab is a human IgGκ monoclonal antibody targeting IL-1β [123]. Rilonacept is a dimeric, glycosylated fusion protein comprising the extracellular domain of IL-1R and the Fc domain of human IgG1. Similar to canakinumab, rilonacept is intended to bind and neutralize IL-1. It binds to both IL-1α and IL-1β with high affinity, and so it has been suggested that rilonacept might have better inhibitory effects in vivo than other IL-1 blockers [123]. Anakinra is a recombinant IL-1Ra that has been shown to be effective for the treatment of a subgroup of AIED patients [97]. Potentially, celastrol, which has several cellular targets—interfering with the production of cytokines, chemokines, and inflammatory mediators such as IL-1β and TNF-α; inhibiting cell invasion and proliferation; and suppressing bone resorption—could constitute a potential candidate for the treatment of inflammatory diseases.
CONCLUSION
All these approved drugs could potentially be beneficial for the treatment of autoimmune/autoinflammatory MD. Since no cure exists for MD, preclinical research and clinical trials repurposing these drugs should be promoted in the next few years. We propose, for instance, to start employing these drugs as secondary treatments. Clinical trials have demonstrated the efficacy of tocilizumab, a humanized anti-IL-6 receptor antibody, for patients with RA and Castleman disease [116], and it may play a role in asthma and other inflammatory pulmonary diseases [124], leading to approval of this innovative drug for the treatment of these diseases. Since IL-6 has been demonstrated to play a significant role in the development of various autoimmune and inflammatory diseases, recruiting MD patients with a history of any of these comorbidities could be a potential starting point to treat them.
HIGHLIGHTS
▪ High levels of interleukin (IL)-1β and tumor necrosis factor-α in some Meniere disease (MD) patients suggest that it is a chronic inflammatory disorder.
▪ The quantitative trait locus rs4947296 regulates the TWEAK/FN14 pathway and nuclear factor-kappa B-mediated inflammation in bilateral MD.
▪ IL-1β, CCL3, CCL22, and CXCL1 may be used as biological markers to distinguish MD and vestibular migraine.
Notes
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conceptualization: all authors. Data curation: LF. Formal analysis: all authors. Funding acquisition: JALE. Methodology: LF. Project administration: JALE. Visualization: all authors. Writing–original draft: LF. Writing–review & editing: all authors.
Acknowledgements
We acknowledge all patients and participants in the Meniere Disease Consortium (MeDiC). Jose Antonio Lopez-Escamez has received funds to support research on the immune response in Meniere disease from Instituto de Salud Carlos III and European Regional Funds (grants no. PI17/1644 and PI20-1126) and the Andalusian Health Department (grants no. PE-0356-2018 and PI027-2020).