INTRODUCTION
Usher syndrome (USH) is an autosomal recessive disorder considered as the most common cause of hereditary deaf-blindness in human, accounting for over 50% of individuals who are both deaf and blind [1]. USH is characterized by bilateral sensorineural hearing loss, progressive visual loss due to retinitis pigmentosa (RP), and vestibular dysfunction. On the basis of the severity and onset of audiovestibular features, USH is differentiated into three types: Usher syndrome type I (USH1) is the most severe form and is manifested by severe to profound congenital deafness, vestibular areflexia, and prepubertal onset of RP. Usher syndrome type II (USH2) is characterized by congenital moderate to severe hearing loss, preserved vestibular function, and RP with an onset at the second decade of life. Usher syndrome type III (USH3) shows progressive hearing loss, RP, and variable vestibular response [2].
USH is clinically and genetically heterogeneous. Total 12 loci and 9 genes thereon have been identified in USH [3]. The mutations of the USH2A have been known to be the most common cause of USH2, accounting for up to 85% of the phenotype [4]. Here we describe a novel mutation identified in the USH2A in a Korean woman with USH2.
CASE REPORT
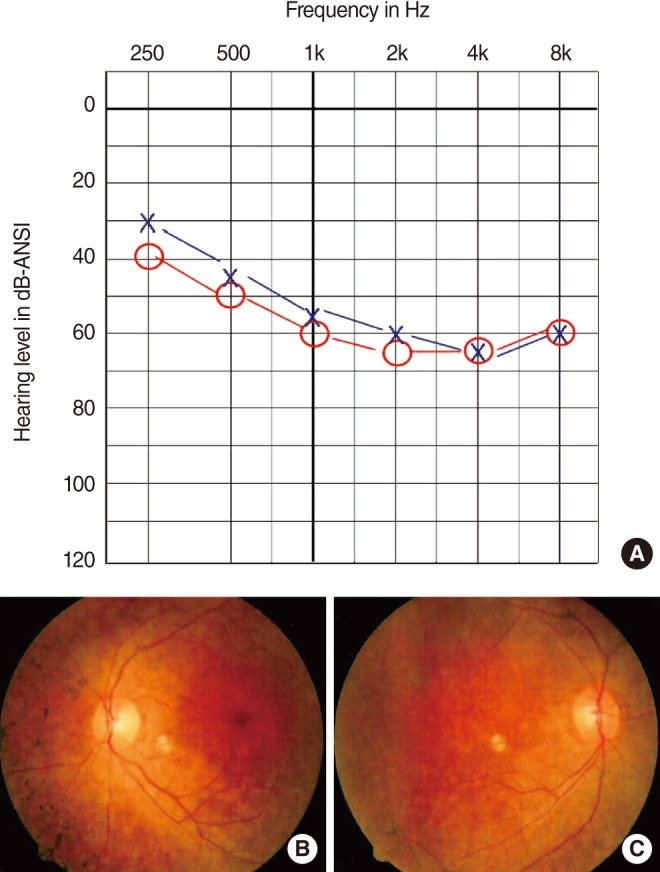
A 34-year-old female was referred to our hospital because of bilateral hearing disturbance since childhood. She complained of a mild hearing difficulty but had not been wearing hearing aids. She had been also suffering from night blindness since adolescence and loss of visual field since age 17. Her visual deterioration had been progressive and she was almost blind at the time of presentation. She did not complain of vertigo. The family history was unremarkable. Physical examination revealed no obvious abnormalities of the external ear or tympanic membrane. For audiologic evaluation, pure tone audiogram (PTA), impedance audiogram, and auditory brainstem evoked response were performed. PTA showed bilateral down-sloping moderate sensorineural hearing loss (Fig. 1A). The threshold of auditory brainstem response was 60 dB HL on both sides. Electronystagmography with a bithermal caloric test demonstrated non specific finding. Temporal bone computed tomography and magnetic resonance imaging showed no specific findings. On ophthalmologic examinations, the lens and the cornea were intact, but the fundoscopic examination revealed bony spicule pigmentation with attenuated retinal vessels in both eyes, the typical finding of RP (Fig. 1B, C). The electroretinogram showed non-detectable waveforms in scotopic and photopic condition in both eyes. Taken together, she was clinically diagnosed as having USH2. The detailed clinical presentation of the patient was previously described by Boo et al. [5].
Molecular genetic analysis
After obtaining written informed consent from the patient, genomic DNA was extracted from peripheral blood leukocytes. We performed polymerase chain reaction (PCR) and direct sequencing of all the 71 coding exons and their flanking intronic sequences of the USH2A using primer pairs designed by the authors. Direct sequencing was performed on the ABI Prism 3100 Genetic Analyzer with the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, USA). Sequence variations were analyzed with reference to the wild type sequence (GenBank accession No. NM_206933.1) using the Sequencher program (Gene Codes Corp., Ann Arbor, MI, USA). For the protein sequence, reference sequence NP_996816.2 was used. The sequence variation detected was described according to the recommendations by the Human Genome Variation Society, having the A of the ATG translation starting codon as +1 at the cDNA level and the corresponding methionine as +1 at the protein level.
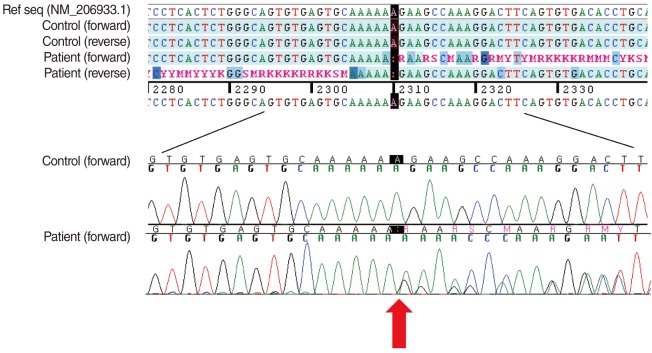
As a result, we detected a heterozygous single nucleotide deletion in the exon 13 of the USH2A, which was expected to result in a frameshift leading to premature termination at the 787th codon (c.2310delA; p.Glu771LysfsX17) (Fig. 2). A review of the literature and the database revealed that this variation was a novel mutation. We also confirmed that the mutation was not observed in 200 control chromosomes.
DISCUSSION
USH2 is known to account for more than half of the Usher syndromes. USH2 could be easily distinguished from USH1 by the severity and audiometric configuration of the hearing loss and by the preserved vestibular function. USH2 is characterized by moderate to severe sensorineural hearing loss with a high frequency sloping configuration. Vestibular response to a caloric stimulus is typically normal. By contrast, the audiograms in USH1 show no detectable hearing across all frequencies, although there may be some residual hearing at the low frequencies.
The USH2A (OMIM 608400) is located in 1q41 and was first described as comprising 21 exons over 259 kb of genomic DNA [6]. However, in up to 40 to 70% of patients with USH2, only a single mutation was detected in these 21 exons of the USH2A gene [7]. The existence of additional exons, suggested by the presence of larger transcripts, was confirmed in 2004 when van Wijk et al. [8] identified 51 novel exons at the 3'-end of the USH2A. A long open reading frame extends from exon 2 to 72, encoding a putative protein of 5,202 amino acids. The functional significance of the long isoform of the USH2A was shown by the presence of pathological mutations in several of the 51 novel exons in patients with USH2. Up to date, more than 70 different mutations of USH2A have been reported in patients with USH2 from various ethnicities, with some founder mutations [9]. Small deletion mutations account for ~26% of all mutations reported. In the present study, we identified a novel small deletion mutation in exon 13 of USH2A (c.2310delA; p.Glu771LysfsX17). We speculate that the frameshift transcripts from the mutant allele in the patient were subjected to nonsense-mediated mRNA decay, leading to a deficiency of USH2A protein, and thereby, the disease phenotype. Of note, we could found only one mutation, suggesting the presence of the other mutation that cannot be detected on direct sequencing, even involving all coding exons and flanking sequences. Baux et al. identified 34 distinct mutations in the USH2A in affected individuals from 25 families with USH2, using the same molecular genetic technique as in our patient. They observed one patient who was homozygous for a mutation, 22 who were compound heterozygous for two different mutations, and 2 patients (8%) who were heterozygous for only one mutation [10]. This study, along with our patient described herein, suggests that there would be other types of USH2A mutations that cannot be detected by direct sequencing. The possibilities include mutations occurring in the promoter region, other intronic regions, or in the 3'- or 5'-untranslated region. Large genomic rearrangement mutations (deletion or insertion/duplication) can also be considered. Indeed, one patient in the Baux et al.'s series had a large deletion mutation involving at least exon 22, confirmed by semiquantitative PCR [10]. Recent studies suggest that USH2A protein is integrated into a protein network formed by other USH-causing proteins [11]. Therefore, another possibility could be that the other mutation might lie in an exon that has not been characterized yet. Finding the other mutation underlying the disease in such patients will give a new insight into the molecular pathogenesis of USH.
In summary, we described a novel heterozygote frameshift mutation in the USH2A in a Korean woman with USH2. This is the first report of a genetically confirmed case of USH in Korea. To better understand the genetic background of USH2 in the Korean population and thereby to establish an optimized strategy of molecular genetic diagnosis and genotype-phenotype correlations, mutation data from more cases are needed.